文章编号:1004-0609(2009)02-0315-07
Ni掺杂MgH2体系解氢性能的机理
周惦武1,张 健1, 2,刘金水2
(1. 湖南大学 汽车车身先进设计制造国家重点实验室,长沙 410082;
2. 湖南大学 材料科学与工程学院,长沙 410082)
摘 要:采用基于密度泛函理论的Dmol 4.1程序包,通过计算移走H原子所需能量及几何、电子结构的改变,对Ni掺杂MgH2体系解氢性能的机理进行探讨。结果表明:Ni替代Mg和创造Mg空位对MgH2体系解氢而言,均发挥有益作用,而形成Mg空位所需能量(6.51 eV)高于Ni替代Mg所需能量(2.12 eV),表明低温下Ni替代Mg对MgH2体系解氢而言更有利,至此NiF2中的Ni替代MgH2中的Mg,有利于加速化学反应NiF2+3MgH2=MgF2+Mg2NiH4向右进行,使结构稳定的MgH2发生转变,生成结构不稳定的Mg2NiH4,这样体系解氢过程不是通过MgH2,而是转变为通过Mg2NiH4进行,因此,Ni掺杂提高了MgH2体系的解氢性能。
关键词:MgH2;Ni掺杂;密度泛函理论;解氢性能
中图分类号:TG 146.2 文献标识码: A
Mechanism of dehydrogenating properties of
Ni doped MgH2 systems
ZHOU Dian-wu1, ZHANG Jian1, 2, LIU Jin-shui2
(31. State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, Hunan University,
Changsha 410082, China;
2. School of Materials Science and Engineering, Hunan University, Changsha 410082, China)
Abstract: According to experimental results in which the dehydrogenating properties of MgH2 systems were improved by addition of NiF2 as catalyst, the energy to remove H atoms, the geometry and electronic structure of MgH2 systems were calculated by using Dmol 4.1 program based on the density functional theory, and the mechanism of improved properties on Ni doped MgH2 systems were also analyzed. The results show that although both Ni substitution and Mg vacancies are effective in desorbing hydrogen at lower temperatures, the substitution of Ni at the Mg site is energetically more favorable than the formation of Mg vacancies. The Ni atoms of NiF2 can replace some Mg atoms of MgH2 systems, the reaction of NiF2+3MgH2=MgF2+Mg2NiH4 during mill process is accelerated, thus, MgH2 with the higher stability can be changed into Mg2NiH4. Because of a ternary hydride Mg2NiH4 with lower stability forming, the dehydrogenating properties on Ni doped MgH2 systems are improved.
Key words: MgH2; Ni doping; density functional theory; dehydrogenating properties
镁氢化合物(MgH2)理论储氢量(质量分数为7.6%)大于国际能源机关(IEA)确定未来新型储氢材料的标准(5.0%),其作为储氢材料进行应用最具诱惑力,然而体系吸放氢性能差,限制了其实际应用。为提高其吸氢性能,人们采用球磨技术替代传统的高温熔炼技术;由于球磨改变了镁颗粒的表面结构,使表面产生较多缺陷,因而球磨法制备的镁纳米晶,比传统镁粉具有更快的吸氢动力学[1?2]。为改善其解氢性能,目前大多数工作集中在向MgH2体系内加入少量催化剂,其中添加3d过渡金属(如Ni、Co、Mn、Cu、Ti、Fe、V等)[3?4]、非3d过渡金属(如Ge、Nb等) [3, 5]、金属间化合物(如LaNi5, FeTi, ZrFe1.4Cr0.6等)[6?8]、金属氧化物(如Nb2O5、Fe3O4等)[9]的实验研究较多。这些催化剂在较高温度下可使MgH2的解氢动力学性能得到一定程度改善。最近JIN等[10]开展了一个较有创造性的研究工作,即在MgH2中加入FeF2、NiF2、TiF3、NbF5、CuF2、VF4、ZrF4、CrF2等金属氟化物作催化剂,结果发现体系显示出优异的解氢性能,催化效果由强到弱的顺序依次为:NiF2、TiF3、VF4、NbF5、FeF2、ZrF4、CrF2、CuF2。
关于MgH2体系解氢性能的理论机制研究,各国学者开展了一些工作。PELLETIER等[9]运用XRD谱,基于对MgH2-5%(摩尔分数)Nb体系吸放氢过程中相结构的转变,对MgH2-Nb体系的解氢机制进行了研究,猜测MgH2-Nb体系优异的解氢性能是由于NbHx、Mg的形核生长在β-NbH/β-MgH2相界上开始进行造成的;SONG等[11]、SHANG等[3]和LI等[12]则采用第一原理计算的方法,通过构建MgH2固溶模型,分别研究了金属元素(如Al、Ti、Fe、Ni、Cu、Nb)和金属氧化物(如Nb2O5)改善MgH2体系解氢性能的催化机理;本文作者所在课题组近期通过构建清洁、空位缺陷Mg(0001)表面吸附氢分子(H2)前后以及Fe合金化MgH2体系的能量与电子结构,探讨了球磨及Fe合金化改善MgH2体系吸放氢性能的微观机理[13],此外为揭示MgH2-V体系的解氢动力学行为,基于对VH0.81相结构的计算,构造VH/MgH2相界模型,研究了该体系解氢能力的电子机制[14]以及体系H原子的吸附与扩散性能[15]。
在JIN等[10]研究的众多金属氟化物中,NiF2催化效果最好,研究意义最大,然而其解氢性能提高的理论机制却不清晰,目前国内外也未见有相关的文献报道。关于NiF2能有效改善MgH2体系的解氢性能,由于JIN等[10]实验发现,高能球磨过程中有MgF2和Mg2NiH4两种产物存在,由此可推测NiF2的加入使体系发生了NiF2+3MgH2=MgF2+Mg2NiH4的化学反应,产物MgF2可能是MgH2中的一部分Mg与NiF3中的F化合形成的,这样MgH2中相应地留下Mg空位,Mg空位有可能影响体系的解氢;而产物Mg2NiH4则可能是NiF3中Ni替代了MgH2中的一部分Mg形成(Mg, Ni)Hx固溶体,再由固溶体接着转变而形成的。Ni替代MgH2中的Mg,可能改变了MgH2局部的几何与电子结构。由此看来,Ni替代MgH2中的Mg、MgH2中形成Mg空位等都有可能对MgH2体系的解氢性能产生较大的影响。基于上述分析和推测,本文作者采用基于密度泛函理论的Dmol 4.1程序包,考察Ni替代MgH2中的Mg和创造Mg空位对MgH2体系中Mg―H、Ni―H键长的变化,计算移走H原子所需能量与体系几何、电子结构的改变,期望对Ni掺杂提高MgH2体系解氢性能的机理有所了解,从而为新型镁基储氢材料的设计提供理论指导。
1 计算模型与方法
为研究MgH2体系几何、电子结构与能量的改变,本研究中构造了由48个原子组成的MgH2(2×2×2)超胞(Mg16H32)模型[12],如图1(a)所示,其中MgH2晶体的晶格常数a=0.450 1 nm,c=0.301 0 nm。空间群为P42/mnm (NO.136)。晶胞中各原子坐标为:+2Mg?(0, 0, 0);+4H?(0.304, 0.304, 0)[11]。为考察Ni替代MgH2中的Mg和创造Mg空位对MgH2体系解氢性能的影响,本研究中分别将Ni原子替代MgH2(2×2×2)超胞(Mg16H32)模型中心的Mg原子和将MgH2超胞中心的Mg原子取走形成Mg空位,构造出相应的Mg15NiH32和Mg15H32超胞模型,分别如图1(b)和1(c)所示。
本研究中计算MgH2体系几何优化、电子结构与能量时,采用的是基于密度泛函理论的Dmol 4.1程序包,电子交换关联能函数采用LDA近似的PWC形 式[16],势函数取全电子位势,电子波函数采用DND基函数[17],布里渊区积分采用Monkhorst-Pack形式的特殊K点方法[18]。能量计算前先进行几何优化(注:超胞晶格常数固定不变,只优化内部原子,以获得其最为稳定的位置),优化时精度设置为:能量≤1.0×10?5 Ha,应力≤0.004 Ha,位移≤0.000 5 nm。在MgH2中创造Mg空位,计算体系在高温下的稳定结构时,采用程序中的Dynamics模块。选取NVT,电子交换关联能函数采用GGA近似的BLYP形式,势函数取全电子位势,电子波函数采用带d轨道的双数值基(DNP) 函数,时间步长取1.0 fs,总模拟时间取0.01 ps,采用Fine网格散点和Smearing energy进行能量快速 收敛。
2 计算结果与分析
2.1 Ni替代Mg对体系解氢的影响
MgH2超胞(Mg16H32)模型几何优化后的结构示意图如图2所示,分析超胞中心Mg(即Mg(0),如图2(b)所示)原子与其第一近邻H (即H(A))原子、第二近邻H(即H(B) )原子之间的键长变化情况,结果如图3所示。由图可知,中心Mg(0)―H(A)之间的键长为0.193 5 nm,Mg(0)― H(B)之间的键长为0.1955 nm。Ni替代中心Mg(0)形成(Mg15Ni)H32超胞模型后,与替代前相比,Ni―H(A)与Ni―H(B)的键长分别为0.165 8 nm和0.171 0 nm,与替代前Mg―H之间的键长相比,Ni―H之间的键长变短,分别减少0.029 7和0.024 5 nm,H原子更靠近Ni原子,在一定程度上,显示Ni原子对H原子有更强的亲和力,Ni―H之间的成键作用强于Mg―H之间的。

图1 3种超胞模型
Fig.1 Three kinds of supercell models: (a) Mg16H32; (b) (Mg15Ni)H32; (c) Mg15H32
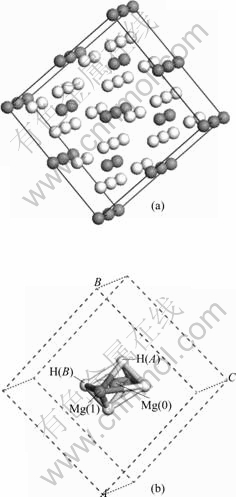
图2 MgH2超胞模型
Fig.2 MgH2 supercell model: (a) MgH2 supercell; (b) Octahedral

图3 H―Mg(Ni)原子之间的键长变化
Fig.3 Bonding distance changes between H and Mg(Ni) atoms
MgH2超胞的态密度如图4(a)所示。由图可见,MgH2在费米能级右边附近存在一个能量间隙,为0.10~0.15 Ha(约0.3~0.4 eV)。费米能级以下的价带部分主要来自Mg(s)与H(s)成键电子的贡献,而以上部分主要是Mg(s)的贡献,H(s)的贡献较少。进一步分析发现,价带部分的Mg(s)与H(s)之间存在一定程度的杂化作用;Ni替代Mg后,态密度如图4(b)所示,能量间隙向低能级移动并且变窄;而在费米能级附近,出现了一个新的能量区间,其主要来自Ni(d)成键电子的贡献(按图4(b)中的比例未显示出来),而在价带部分,Ni(s)、Ni(d)与H(s)之间存在强烈的杂化作用。
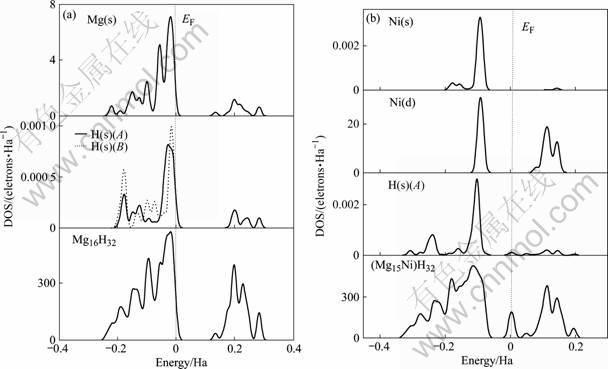
图4 Ni替代Mg前后MgH2的态密度
Fig.4 DOS of MgH2 systems: (a) Before Ni atom substituting Mg atom; (b) After Ni atom substituting Mg atom
为研究Ni替代Mg对体系解氢的影响,将MgH2超胞(Mg16H32)中心Mg(0)第一近邻的H(H(A))原子移走,结果发现第二近邻H(H(B))原子与Mg(0)之间的键长与未移走H(A)原子前的相比,变化不大(如图5所示),均为0.195 5 nm;而Ni替代MgH2中的Mg后,H(B)―Ni之间的键长为0.167 0 nm,与未移走H(A)原子(0.171 0 nm)相比,却明显变短,这也一定程度上表明,Ni与H之间的成键作用较强,Ni类似“磁铁石”,移走第一近邻H原子,还能将周围第二近邻、甚至更远的H原子源源不断地强烈吸引到自身周围,并有望形成NiHx集团。

图5 H(B)―Mg(0)(Ni)原子之间的键长变化
Fig.5 Bonding distance changes between H(B)―Mg(0)(Ni) atoms
进一步采用下式计算从MgH2体系移走H原子所需要的能量[19]:
Ecoh=E(Mg16?xNixH32?n)?E(Mg16?xNixH32)+n/2E(H2) (1)
式中 x和n分别表示体系中Ni和H原子的个数。计算所需要的能量如图6所示。发现Ni替代Mg后,MgH2体系中移走1个H(即第一近邻H(A))原子所需要的能量为0.75 eV,明显低于未替代前的1.77 eV,而移走2个H原子(即第一近邻H(A)和第二近邻H(B)),发现Ni替代Mg,所需能量(1.61 eV)仍低于未替代前(1.90 eV)的情形。由此可看出:Ni替代Mg对MgH2体系解氢是有利的。

图6 MgH2移走H原子所需要的能量
Fig.6 Energy for removing H atoms from MgH2 systems
2.2 Mg空位对体系解氢的影响
JIN等[10]指出,NiF2能有效改善MgH2体系的解氢性能,这与实验中高能球磨产生的高密度缺陷有关,但其对这些缺陷的作用本质却没有深入研究。此外,高能球磨MgH2与NiF2的过程中形成了副产物MgF2,而MgF2的形成也可能与MgH2中形成的Mg空位密切相关。为此,探讨Mg空位对体系解氢的影响很有必要。在MgH2体系中创造1个Mg空位,H―H之间的键长的变化情况如表1所示。
表1 创造1个Mg空位前后MgH2体系中H―H键长的变化情况
Table 1 Bonding distance changes of H―H atoms in MgH2 systems with and without Mg vacancy
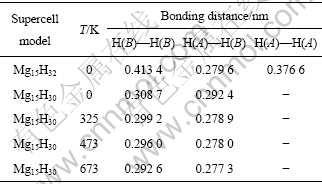
从表1可看出:0 K时,Mg空位的存在,MgH2体系中H―H之间的键长最短为0.279 6 nm,与H2分子中H―H之间的键长(0.074 1 nm)相比,表明体系H2分子未形成,移走2个H原子(即第一近邻H(A)和第二近邻H(B)),H―H之间的最短键长为0.292 4 nm,H2分子仍未形成;升高体系温度到325 K时,发现与0K时比较,H―H之间的最短键长为0.278 9 nm,虽然仍大于H2分子中H―H之间的键长,但形成H2分子的倾向却在增强。由于通常MgH2体系真实的实验温度为473~673 K,为此比较了H―H之间的键长在真实实验温度范围内的变化情况,如图7所示。从图7可见,体系在473和673 K时,H―H之间的最短键长分别为0.278 0 nm和0.277 3 nm,仍大于H2分子中H―H之间的键长(0.0741 nm),但温度进一步升高却导致H―H键长继续缩短,表明创造Mg空位后,移走2个H原子,MgH2体系形成H2分子的倾向在增强,Mg空位的存在利于提高MgH2体系的热力学行为。
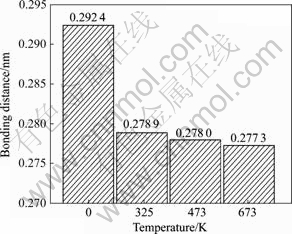
图7 不同温度下H―H原子之间的键长的变化情况
Fig.7 Bonding distance changes of H―H atoms at different temperatures
为进一步证实创造Mg空位后对MgH2体系解氢产生有益的作用,采用式(1)计算了MgH2体系移走2个H原子所需要的能量,发现计算结果为负,其值为2.76 eV,而未形成Mg空位前所需能量为正,由此可看出,Mg空位的存在,对MgH2体系解氢也是十分有利的[19]。
2.3 Ni掺杂体系解氢性能提高的机理
由2.1和2.2节部分可看出,Ni替代Mg和创造Mg空位,均对MgH2体系解氢发挥十分有益的作用,但低温下两者之中到底哪种作用占优则不明确,为进一步研究这个问题,本研究中分别采用如下公式计算了Ni替代Mg和形成Mg空位所需要的能量[20]:
H1=[E(Mg15NiH32)+E(Mg)]?[E((Ni)+E(Mg16H32)) (2)
H2=E(Mg15H32)+E(Mg)?E(Mg16H32) (3)
计算结果发现,形成Mg空位需要的能量为6.51 eV,明显高于Ni替代Mg所需要的能量2.12 eV,因此,低温下Ni替代Mg与形成Mg空位相比,对MgH2体系解氢而言发挥更大作用。这样对NiF2催化MgH2体系解氢而言,低温下,主要是NiF2中的Ni替代MgH2中的Mg,加速了化学反应NiF2+3MgH2=MgF2+ Mg2NiH4向右进行,使MgH2转变生成了Mg2NiH4。对2.1节中Ni替代Mg后分析发现,Ni(s)、Ni(d)与H(s)之间存在强烈的杂化作用,移走第一近邻H原子后,Ni与第二近邻、甚至更远的H原子成键作用更强,Ni将H强烈吸引到周围,有形成NiHx集团的趋势,这样Ni替代Mg使MgH2容易转变为(Mg, Ni)Hx固溶体,而(Mg, Ni)Hx固溶体是解氢的通道[10],因此,有望形成Mg2NiH4;JIN等[10]实验观察NiF2催化MgH2体系中,存在MgF2和Mg2NiH4两种产物,证实了Ni替代Mg对MgH2体系解氢有利的分析结果。
进一步分析可知,MgH2的合金形成热的实验值为(?76.15±9.2)kJ/mol[21],相结构稳定,体系不容易解氢,而Mg2NiH4合金形成热的实验值为?62.7 kJ/mol[21],与MgH2相比较而言,Mg2NiH4相结构不稳定,体系解氢容易。这样体系解氢过程不是通过MgH2,而是转化为通过Mg2NiH4来进行,因此,Ni掺杂提高了MgH2体系的解氢性能。
3 结论
1) Ni替代Mg和创造Mg空位,均对MgH2体系解氢发挥十分有益的作用,其中低温下,Ni替代Mg发挥的作用更大。
2) NiF2中的Ni替代MgH2中的Mg,有利于加速化学反应NiF2+3MgH2=MgF2+Mg2NiH4向右进行,使结构稳定的MgH2发生转变生成了结构不稳定的Mg2NiH4,体系解氢过程不是通过MgH2,而是通过Mg2NiH4进行,因此,Ni掺杂提高了MgH2体系的解氢性能。
REFERENCES
[1] ZALUSKA A, ZALUSKI L, STR?M-OLSEN J O. Nanocrystalline magnesium for hydrogen storage[J]. J Alloys Compd, 1999, 288(1/2): 217?225.
[2] JURCZYK M, SMARDZ L, OKONSKA I, JANKOWSKA E, NOWAK M, SMARDZ K. Nanoscale Mg-based materials for hydrogen storage[J]. Int J Hydrogen Energy, 2008, 33(1): 374?380.
[3] SHANG C X, BOUOUDINA M, SONG Y, GUO Z X. Mechanical alloying and electronic simulations of (MgH2+M) systems (M=Al, Ti, Fe, Ni, Cu and Nb) for hydrogen storage[J]. Int J Hydrogen Energy, 2004, 29(1): 73?80.
[4] HUOT J, HAYAKAWA H, AKIBA E. Preparation of the hydrides Mg2FeH6 and Mg2CoH5 by mechanical alloying followed by sintering[J]. J Alloys Compd, 1997, 248(1/2): 164?167.
[5] GENNARI F C, CASTRO F J, URRETAVIZCAYA G, MEYER G. Catalytic effect of Ge on hydrogen desorption from MgH2[J]. J Alloys Compd, 2002, 334(1/2): 277?284.
[6] LIANG G, HUOT J, BOILY S, VAN NESTE A, SCHULZ R. Hydrogen storage in mechanically milled Mg-LaNi5 and MgH2-LaNi5 composites[J]. J Alloys Compd, 2000, 297(1/2): 261?265.
[7] MANDAL P, DUTTA K, RAMAKRISHNA K, SAPRU K, SRIVASTAVA O N. Synthesis, characterization and hydrogenation behaviour of Mg-xwt.%FeTi (Mn) and La2Mg17-xwt.%LaNi5 new hydrogen storage composite alloys[J]. J Alloys Compd, 1992, 184(1): 1?9.
[8] WANG P, WANG A M, DING B Z, HU Z Q. Mg-FeTi1.2(amorphous) composite for hydrogen storage[J]. J Alloys Compd, 2002, 334(1/2): 243?248.
[9] PELLETIER J F, HUOT J, SUTTON M, SCHULZ R, SANDY A R, LURIO L B, MOCHRIE S G J. Hydrogen desorption mechanism in MgH2-Nb nanocomposites[J]. Phys Rev B, 2001, 63: 052103?052106.
[10] JIN S A, SHIM J H, CHO Y W, YI K W. Dehydrogenation and hydrogenation characteristics of MgH2 with transition metal fluorides[J]. J Power Sources, 2007, 172(1/2): 859?862.
[11] SONG Y, GUO Z X, YANG R. Influence of selected alloying elements on the stability of magnesium dihydride. for hydrogen storage applications: A first-principles investigation[J]. Phys Rev B, 2004, 69: 094205?094215.
[12] LI S, JENA P, AHUJA R. Dehydrogenation mechanism in catalyst-activated MgH2[J]. Phys Rev B, 2006, 74: 132106?132109.
[13] 周惦武, 张 健, 刘金水, 彭 平. 球磨条件下Fe合金化改善MgH2体系性能的机理[J]. 中国有色金属学报, 2008, 18(12): 2233?2244.
ZHOU Dian-wu, ZHENG Jian, LIU Jin-shui, PENG Ping. Mechanism of improved properties of MgH2 systems with ball milling and iron addition[J]. The Chinese Journal of Nonferrous Metals, 2008, 18(12): 2233?2244.
[14] ZHOU Dian-wu, PENG Ping, LIU Jin-shui. First-principles calculation of dehydrogenating properties of MgH2-V systems[J]. Science in China: Series E, 2006, 49(2): 129?136.
[15] ZHOU Dian-wu, LIU Jin-shui, PENG Ping. Study on H atoms diffusion and adsorption properties of MgH2-V systems[J]. Science in China: Series E, 2008, 51(7): 979?988.
[16] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77(18/28): 3865?3868.
[17] PACK J D. MONKHORST H J. Special points for Brillouin-zone integrations―A reply[J]. Phys Rev B, 1977, 16(4/15): 1748?1749.
[18] DELLEY B. Analytic energy derivatives in the numerical local-density-functional approach[J]. J Chem Phys, 1991, 94(11): 7245?7250.
[19] MEDVEDEVA M I, GORNOSTYREV Y N, NOVIKOV D L, MRYASOV O N, FREEMAN A J. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl[J]. Acta Mater, 1998, 46(1/2): 3433?3442.
[20] SAHU B R. Electronic structure and bonding of ultralight LiMg[J]. Mater Sci Eng B, 1997, 49(1/2): 74?78.
[21] BOGDANOV?? B, BOHMHAMMEL K, CHRIST B, REISER A, SCHLICHITE K. Thermodynamic investigation of the magnesium-hydrogen system[J]. J Alloys Compd, 1999, 282(1/2): 84?92.
基金项目:教育部博士点专项科研基金资助项目(200805321032);湖南省科技计划资助项目(2008GK3083);教育部长江学者与创新团队发展计划项目(531105050037)
收稿日期:2008-06-20;修订日期:2008-09-26
通讯作者:周惦武,副教授,博士;电话:13017297124;E-mail: ZDWe_mail@yahoo.com.cn
(编辑 何学锋)

