网络首发时间: 2017-06-19 17:50
稀有金属 2018,42(07),705-712 DOI:10.13373/j.cnki.cjrm.xy17040032
影响Nb替位掺杂γ-TiAl基合金延性的电子性质因素
宋庆功 赵俊普 顾威风 甄丹丹 郭艳蕊 李泽朋
中国民航大学理学院低维材料与技术研究所
中国民航大学中欧航空工程师学院
摘 要:
采用基于密度泛函理论的第一性原理方法, 计算研究了影响Nb替位Ti (或Al) 掺杂, 且浓度为1.85%, 2.78%, 4.17%, 6.25% (原子分数, 下同) 的γ-Ti Al合金延性的电子性质因素。结果显示, Nb替位掺杂具有较好的能量稳定性, 且不同掺杂浓度引起的晶格参量的改变不同。当Nb替代Al原子且掺杂浓度为1.85%4.17%时, 合金的轴比将会减小更接近于1, 此时合金的延性改善效果明显, 就所研究的晶胞模型而言, Ti12Al11Nb合金的延性最佳。故以掺杂浓度为4.17%的Ti12Al11Nb和Ti11Nb Al12合金为例, 研究掺杂Nb改善γ-Ti Al基合金延性的电子结构因素。研究结果表明:在该掺杂浓度情况下, Nb替代Al原子使Ti 4s→3d, Al 3s→3p的电荷转移明显减少, Al p-Ti d轨道杂化作用减弱, 形成金属键的自由电子数量增多, 金属性增强, 合金延性得到改善;Nb替代Al原子掺杂会使合金的共价性减弱, 且化学键Al-Al, Ti-Ti和Al-Ti强度明显趋同, 这将会显著提高γ-Ti Al基合金的各向同性程度, 从而使该类合金的延性得以改善。
关键词:
Nb掺杂γ-TiAl;晶体结构;延性;电子性质;
中图分类号: TG146.23
作者简介:宋庆功 (1958-) , 男, 河北唐山人, 博士, 教授, 研究方向:航空材料、金属间化合物;电话:022-24092510;E-mail:qgsong@cauc.edu.cn;
收稿日期:2017-04-19
基金:国家自然科学基金项目 (11304380);中国民航大学中央高校基本科研业务费 (3122014K001) 资助;
Electronic Properties Influencing Ductility of Nb-Substituted γ-TiAl Based Alloys
Song Qinggong Zhao Junpu Gu Weifeng Zhen DANDan Guo Yanrui Li Zepeng
Institute of Low Dimensional Materials and Technology, College of Science, Civil Aviation University of China
Sino-European Institute of Aviation Engineering, Civil Aviation University of China
Abstract:
Nb-dopedγ-Ti Al based alloys were investigated using first principles method based on density functional theory.The crystal structure stability, ductility and electronic properties were calculated in four different impurities:1.85%, 2.78%, 4.17%and6.25% (atomic fraction) .The results showed that all Nb-doped alloys got a good energy stability, which indicated those can be prepared in experiment and stable in existence.The lattice parameter changed with Nb doping concentration.When Nb substituted Al atom by 1.85%4.17%, the axial ratio of the alloy would decrease to 1.Then the ductility of the alloy was improved obviously.The ductility of Ti12Al11Nb alloy was the best among all super cell models.Take Ti12Al11Nb and Ti11Nb Al12alloys that doped with 4.17%as examples, the electronic structure factors of improving the ductility of Nb-dopedγ-Ti Al alloys were studied.The results showed that under this doped concentration conditions, there were two electronic structure factors for the improvement of ductility.Firstly, thanks to that the transfer charge of Ti 4s→3d and Al 3s→3p decreased significantly, the p-d orbital hybridization were weakened, the number of free electron metal bond were added, metal property were strengthened, so the ductility ofγ-Ti Al based alloys were enhanced.Secondly, the strength difference degree among of Al-Al, Ti-Ti and Al-Ti bonds reduced significantly, which improved the isotropic degree ofγ-Ti Al based alloys remarkably.In summary, this change of electronic structure ofγ-Ti Al would improve its ductility.
Keyword:
Nb-doped γ-TiAl; crystal structure; ductility; electronic property;
Received: 2017-04-19
Ti Al基合金因密度低、比强度高以及优良的高温性能而成为航空航天、汽车工业等领域的新一代备用材料[1,2]。但其室温延展性较差、加工成型困难严重制约着该类合金材料的工业化生产和在航空航天、能源动力、交通运输等领域中的应用[3,4]。添加合金元素是改善该类合金性能的一条有效途径[5,6]。例如, 添加12.5% (原子分数, 下同) 的Mo元素既可以改善γ-Ti Al合金的延性, 也能提高其稳定性和强度[7]。其原因在于, Mo和Ti原子发生强烈的s-s, p-p, d-d电子相互作用, 并且随着掺杂浓度的增大, 电子相互作用增强, 有效地束缚了合金中Ti和Al原子的迁移, 有助于提高合金的稳定性和强度[7];添加4.17%的La, 能有效减小位错移动的阻力, 从而使γ-Ti Al合金的延性得到改善[8];添加3.125%的Y, 有助于削弱Ti d-Al p的方向性, 增强Ti-Ti和Al-Al的键合强度, 从而使γ-Ti Al合金脆性得到缓解[9];添加Fe或Co可以改善Ti Al合金的延性, 添加Ni则因形成Ni Ti相而使该合金延性变差[10]。
Nb作为合金化元素, 不仅可明显降低Ti Al合金高温氧化增重, 而且还可以有效改善其力学性能[11]。Nb能有效地提高γ-Ti Al的室温和高温下的性能已经被多次[12,13,14]证明。例如:在实验方面, 李海昭等[15]对Ti48AlxNb2.5V1.0Cr合金 (x=2%, 4%, 7%, 9%) 的延性进行了研究, 结论显示, γ相的成分随Nb浓度增加而增多, 且Nb掺杂浓度为2%和9%时, 合金的延伸率分别为1.2%与0.3%。张兰芝等[16]用正电子湮没谱研究了Nb在Ti Al合金中的掺杂效应, 并得出结论, 低浓度掺杂时Nb原子主要偏聚在合金晶界处, 提高了晶界位置的自由电子密度有利于改善合金的室温韧性;而较高含量的Nb掺杂时, 由于形成了新的晶体结构, 合金基体及晶界处的自由电子密度减少, 导致合金的脆性增加。理论方面, 党宏丽等[17]利用密度泛函理论框架下的第一性原理离散变分 (DV) 和DMol方法研究了Nb元素在γ-Ti Al中合金化效应。结果表明, Nb可以提高杂质原子与其近邻基体原子之间的相互作用进而引起固溶强化效应。吴红丽等[18]对Nb掺杂Ti Al基合金的键合特征进行了研究, 结果表明, 掺杂Nb不仅使Ti O2的生成能垒增大了, 而且使Al2O3的生成能垒减小了, 即有利于促进抗氧化膜Al2O3的形成, 在一定程度上改善Ti Al的抗氧化性能。可见, 杂质Nb对于Ti Al基合金体系的性质有重要影响。
Ti Al合金的相成分实为金属间化合物, 此类材料的性质与其电子结构密切相关[19,20,21]。例如, Ti Al晶体中存在Al p-Ti d方向性键合, 使材料的剪切强度提高, 进而造成其室温延性较差[19];Ti Al合金中电荷分布的各向异性是造成Ti Al合金室温脆性的本质原因[22]。但仍需要从实验上研究确认合金性质对电子结构的依赖关系及其微观机制。最近, 计算模拟方法在新材料设计方面的作用得到了高度肯定[23]。可以说, 材料研究已经进入以前瞻性结构设计和性能预测为前端, 以实验制备研发、性能验证为后端的新型范式, 这对于研发新型材料具有重要作用[8,24]。为了探索Nb改善Ti Al基合金性质的内在电子性质因素, 本文对Nb替位掺杂γ-Ti Al体系进行结构和性质研究。探求改善该类合金延性的途径以及微观机制, 为改善该类合金的综合性质提供理论依据。
1计算模型与方法
1.1结构模型
γ-Ti Al为L10型面心四方结构, 如图1 (a) 所示, 对应的晶格参量a0=b0=0.398 nm, c0=0.404 nm, α=β=γ=90°[25]。其最小结构单元如图1 (b) 所示, 对应的晶格参量a=0.283 nm, c=0.407 nm。
为选择延性改善效果最优的Nb掺杂比例, 本文在最小结构单元的基础上, 构造了2×2×2, 3×2×2, 3×3×2和3×3×3的γ-Ti Al超胞。在上述超胞中, 均掺入1个Nb原子, 形成掺杂体系:Ti8Al7Nb, Ti12Al11Nb, Ti18Al17Nb, Ti27Al26Nb, 即Ti Al1-xNbx系列合金体系和Ti7Nb Al8, Ti11Nb Al12, Ti17Nb Al18, Ti26Nb Al27, 即Ti1-xNbxAl系列合金体系 (x=6.25, 4.17, 2.78, 1.85%) 。图2给出Nb替代Ti原子掺杂得到的4种结构模型。
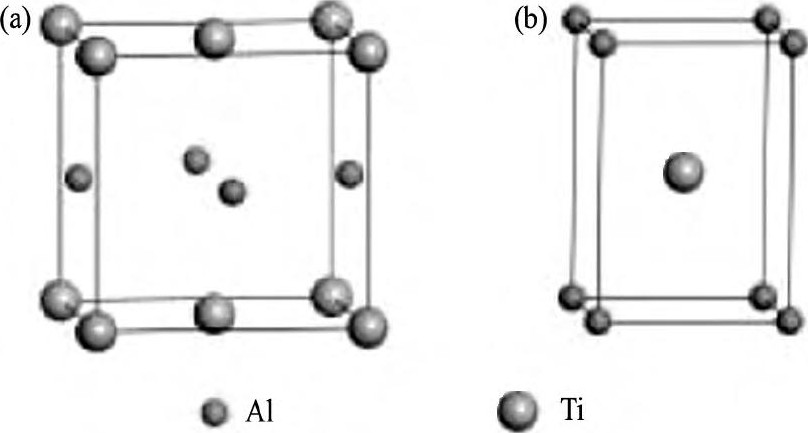
图1 γ-Ti Al结构模型Fig.1 Structure models ofγ-Ti Al
(a) L10unit cell ofγ-Ti Al; (b) Simple tetragonal unit ofγ-Ti Al
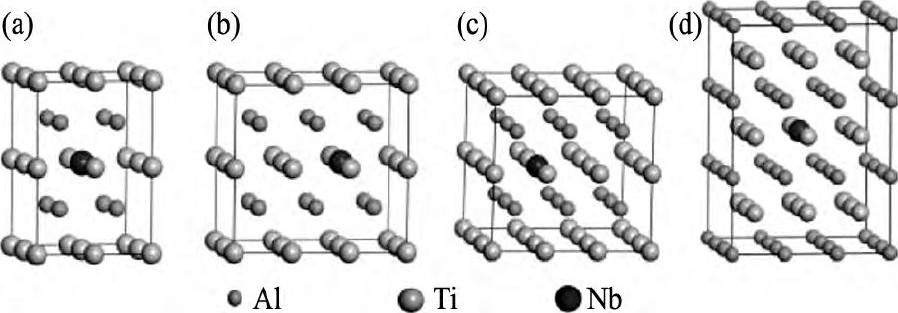
图2 掺杂γ-Ti Al体系的结构模型Fig.2 Structure models of dopedγ-Ti Al systems (a) Ti7Nb Al8; (b) Ti11Nb Al12; (c) Ti17Nb Al18; (d) Ti26Nb Al27
1.2计算方法
本文采用基于密度泛函理论的第一性原理方法结合其它物理与化学理论方法, 利用高性能计算机集群, 在Materials Studio 6.0软件包下的CASTEP (Cambridge serial total energy package) 模块建立晶胞模型, 并完成相关理论计算。首先对纯γ-Ti Al体系进行拟牛顿算法几何优化, 并与相应实验研究结果相对比, 再选取相对误差较小的计算方案。最终方案如下:交换相关能选用广义梯度近似基础下的PBE (Perdew, Burke and Ernzerhof) ;离子实与价电子之间的相互作用采用超软赝势来描述。平面波截断能为320 e V。为使各体系计算所得结果精度一致, 布里渊区k点随掺杂浓度的增加分别设置为:3×3×2 (1.85%) , 3×3×3 (2.78%) , 3×4×3 (4.17%) 以及4×4×3 (6.25%) 。
通过几何优化得到稳定的晶胞模型, 在此基础上, 对合金的几何结构、形成能、布居数、电荷密度、电子态密度等性质进行计算研究与分析。
2结果与讨论
2.1形成能与稳定性
材料的能量稳定性可以用其原子平均形成能的高低进行描述。即原子平均形成能越低, 则该材料的能量稳定性越好[26]。对于Nb掺杂γ-Ti Al基合金体系, 原子平均形成能Ef可表示为:
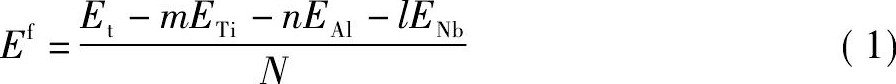
式中, Et表示晶胞的总能量;m, n, l分别表示各个元素在晶胞中的原子个数;N表示晶胞中原子总数;ETi为Ti在单质情况下完全弛豫后的单原子能量 (EAl, ENb同理) , 采用与计算晶胞同样的方案计算得到ETi, EAl, ENb的值分别为-1603.0622, -56.4180, -1551.6089 e V。经计算各个体系的总能量和平均形成能如表1所示。
由表1可知, 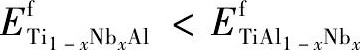 , 即Ti1-x NbxAl系列合金的能量稳定性高于Ti Al1-xNbx。这表明Nb原子替位掺杂时, 占位Ti原子格点的概率大于占据Al原子格点的概率。这种现象可用置换固溶规律进行解释, 即Nb倾向于占据Ti原子格点, 原因在于二者的原子共价半径 (rNb=0.137nm, rTi=0.136 nm) 比较接近, 掺杂引起的晶格畸变较小;相反, Al原子半径 (rAl=0.118 nm) 较小, Nb占据Al原子格点时, 会引起较大的晶格畸变, 使体系的晶格能量升高, 稳定性降低。
, 即Ti1-x NbxAl系列合金的能量稳定性高于Ti Al1-xNbx。这表明Nb原子替位掺杂时, 占位Ti原子格点的概率大于占据Al原子格点的概率。这种现象可用置换固溶规律进行解释, 即Nb倾向于占据Ti原子格点, 原因在于二者的原子共价半径 (rNb=0.137nm, rTi=0.136 nm) 比较接近, 掺杂引起的晶格畸变较小;相反, Al原子半径 (rAl=0.118 nm) 较小, Nb占据Al原子格点时, 会引起较大的晶格畸变, 使体系的晶格能量升高, 稳定性降低。
所有体系的原子平均形成能均为负值, 表明它们均具有较好的能量稳定性, 即在一定条件下Ti Al1-xNbx, Ti1-xNbxAl (x=1.85%, 2.78%, 4.17%, 6.25%, 原子分数) 系列合金是可以实验制备并能稳定存在的。
2.2几何结构与延性
通过对纯γ-Ti Al, Ti Al1-xNbx, Ti1-xNbxAl (x=1.85%, 2.78%, 4.17%, 6.25%;原子分数) 合金进行几何优化, 得到它们的最优结构。为了便于对比分析, 将各个体系最优结构的晶格参量换算成γ-Ti Al最小结构单元对应的量值。利用晶格参量可得其轴比R值 (R为折合成L10结构的量值) 。各个体系的晶格参量与轴比参见表2。为了便于直观地观察它们的变化趋势, 将晶格参量与轴比值绘于图3。
结合表2与图3, 容易看出掺杂体系的晶格参量a均较纯γ-Ti Al增大;除Ti7Nb Al8外, 其余体系的晶格参量c均较纯γ-Ti Al有所降低。由于晶格参量a, c的发生上述变化, 导致掺杂体系的轴比值均较纯γ-Ti Al有所降低。其中, Ti Al1-xNbx系列合金的轴比值随着Nb浓度x (1.85%≤x≤6.25%, 同下) 的增加出现先增加后减小的趋势。Ti1-x NbxAl系列合金的轴比值随着Nb浓度x的增加而增加。
表1 Nb掺杂γ-Ti Al基合金的能量性质Table 1 Energy properties of Nb dopedγ-Ti Al alloys 下载原图

表1 Nb掺杂γ-Ti Al基合金的能量性质Table 1 Energy properties of Nb dopedγ-Ti Al alloys
表2 Nb掺杂掺杂γ-Ti Al合金的几何性质Table 2 Geometrical properties of Nb dopedγ-Ti Al alloys 下载原图
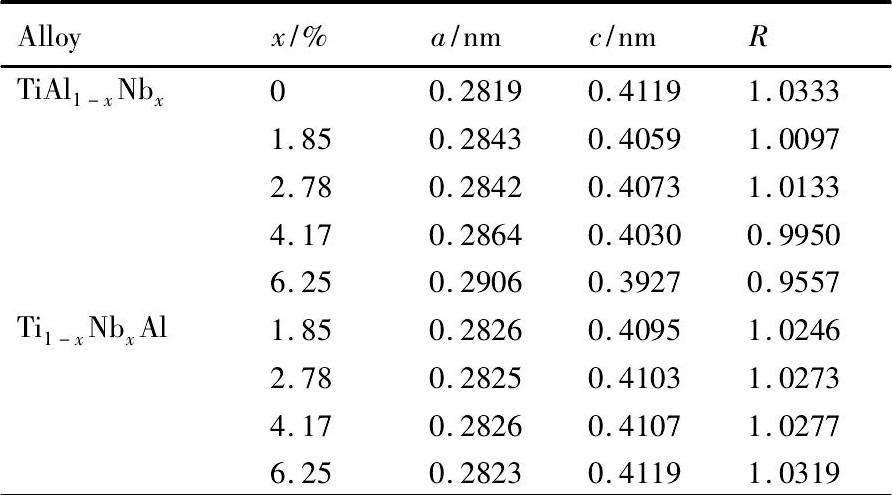
表2 Nb掺杂掺杂γ-Ti Al合金的几何性质Table 2 Geometrical properties of Nb dopedγ-Ti Al alloys
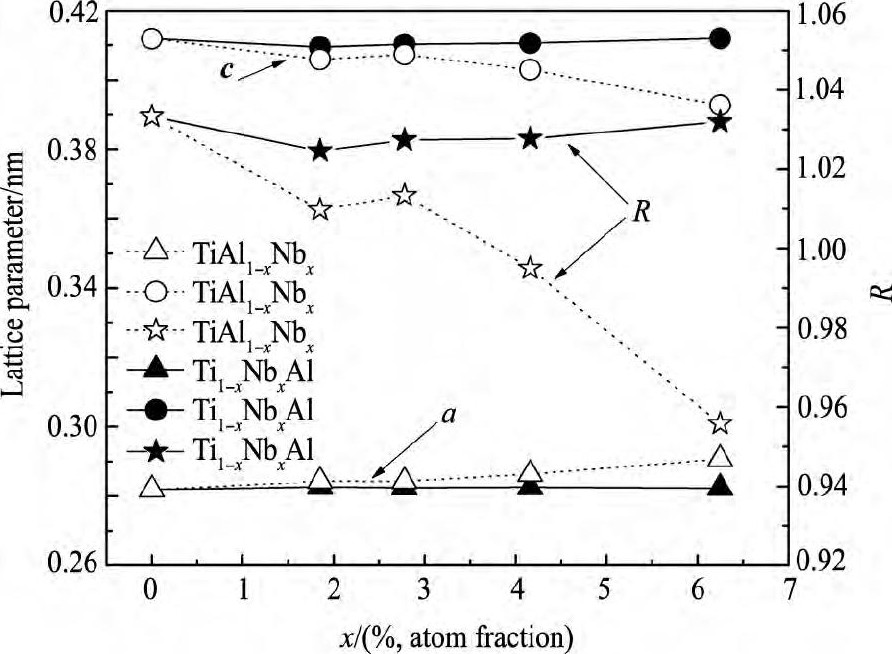
图3 晶格参量与轴比Fig.3 Lattice parameter and their axis ratio
Kawabata等[27]研究发现γ-Ti Al基合金的延性可以用轴比R表征。轴比R越接近于1, 材料的立方度就越好, 这样就可使晶体的对称性和变形协调性得到提高, 并且还能增加滑移系数目, 从而使该类合金的延性得到改善。本文所研究的Nb掺杂体系轴比值除Ti8Al7Nb外, 其余均较纯γ-Ti Al更接近1。这说明它们的延性均较纯γ-Ti Al有所改善。特别是Ti Al1-xNbx系列合金当Nb浓度在1.85%~4.17%时, 最为接近1。其中当Nb浓度为2.78%时, 轴比值大于1 (R=1.0133) ;当Nb浓度为4.17%时, 轴比值小于1 (R=0.9950) 。即可能存在一个Nb浓度x0 (2.78%≤x0≤4.17%) , 使其轴比为1。此时合金Ti Al1-x0Nbx0为立方结构, 该结构各向同性程度最高, 超位错和普通位错之间的差异最小, 有利于位错的移动, 即Ti Al1-x0Nbx0合金的延性较纯γ-Ti Al体系显著改善。就所研究掺杂合金而言, Ti12Al11Nb的轴比值为0.9950, 比其他掺杂合金更接近1, 即其延性最佳。
由图3可知, 当Nb含量较大时, 轴比会远离1, 由此说明体系的延性会下降, 关于这一点, 很多实验已经证实了室温下高铌γ-Ti Al合金的延性较差, 通常会通过后续热处理来改善这一缺陷, 相反的是, 高铌γ-Ti Al合金在高温下的延展性等性质优良, 适合作为一种新型的高温结构材料。
2.3电荷布居数与延性
Al, Ti和Nb原子的价电子组态分别为3s23p1, 3s23p63d24s2和4s24p64d45s1。Ti原子中有2个未配对的3d电子, Al原子也有1个未配对的3p电子, 它们均有很强的局域性。当多价Al原子与Ti原子成键时, Al原子的3p电子与Ti的3d电子将发生轨道杂化, 形成p-d共价键, 导致合金中参与形成金属键的自由电子数量减少[28]。党宏丽等[17]研究表明, 在纯γ-Ti Al合金中, 有电荷从Al原子的3s轨道转移到Al原子的3p轨道 (Al 3s→3p) ;也有电荷从Ti原子的4s轨道转移到Ti原子的3d轨道 (Ti 4s→3d) 。这会直接导致γ-Ti Al合金基质中p-d轨道杂化作用增强、自由电子数量减少, 金属键减弱, 宏观上γ-Ti Al基合金的金属性减弱, 延性降低。该观点也可与Morinaga等[19]研究结论相互佐证。
为了研究γ-Ti Al基合金中Ti, Al原子的电荷转移情况, 表3给出Ti12Al12, Ti12Al11Nb和Ti11Nb Al12合金体系的Mulliken电荷布居数。由表3易知, 合金中原子的电荷均发生了轨道转移, 其中纯γ-Ti Al体系中电荷转移情况与党宏丽等[17]研究结论一致。根据表4可得Al 3s→3p, Ti 4s→3d与Nb 5s→4d的具体电荷转移量, 如图4所示。
为便于表达, 设轨道之间的电荷转移量为Q, 即 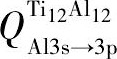 表示Ti12Al12合金中Al原子3s轨道转移到Al原子3p轨道的电荷量 (其他合金原子的电荷转移量同理) 。由图4可知,
表示Ti12Al12合金中Al原子3s轨道转移到Al原子3p轨道的电荷量 (其他合金原子的电荷转移量同理) 。由图4可知, 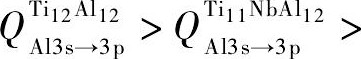
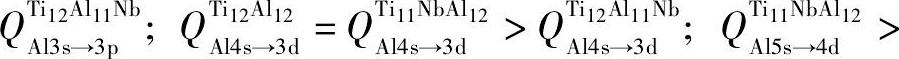
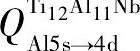 。即Ti12Al11Nb合金中各原子的电荷转移量均低于Ti12Al12和Ti11Nb Al12合金。这将导致Ti12Al11Nb中的p-d轨道杂化作用低于其他两者。因而使Ti12Al11Nb合金中形成金属键的自由电子数量比Ti12Al12和Ti11Nb Al12合金多。即Ti12Al11Nb合金的金属性与延性均高于Ti12Al12和Ti11Nb Al12合金。费米能级附近的电子态密度 (DOS) 亦可佐证上述结论。图5给出Ti12Al12, Ti11Nb Al12和Ti12Al11Nb合金的Al-p轨道与Ti-d轨道的电子态密度图。
。即Ti12Al11Nb合金中各原子的电荷转移量均低于Ti12Al12和Ti11Nb Al12合金。这将导致Ti12Al11Nb中的p-d轨道杂化作用低于其他两者。因而使Ti12Al11Nb合金中形成金属键的自由电子数量比Ti12Al12和Ti11Nb Al12合金多。即Ti12Al11Nb合金的金属性与延性均高于Ti12Al12和Ti11Nb Al12合金。费米能级附近的电子态密度 (DOS) 亦可佐证上述结论。图5给出Ti12Al12, Ti11Nb Al12和Ti12Al11Nb合金的Al-p轨道与Ti-d轨道的电子态密度图。
表3 γ-Ti Al基合金的Mulliken电荷布居数Table 3 Mulliken population ofγ-Ti Al based alloys 下载原图

表3 γ-Ti Al基合金的Mulliken电荷布居数Table 3 Mulliken population ofγ-Ti Al based alloys

图4 电荷转移量Fig.4 Transfer charge ofγ-Ti Al based alloys
由图5可知, 3种合金Al-p与Ti-d轨道的电子态密度大小关系均为Ti12Al12>Ti11Nb Al12>Ti12Al11Nb, 即可参与形成p-d共价键的自由电子数量关系也为Ti12Al12>Ti11Nb Al12>Ti12Al11Nb。所得结论与电荷布居数给出的结论一致。
2.4重叠布居数与延性
合金的性质与其内部化学键的性质密切相关。原子间的重叠布居数可以描述成键原子之间的相互作用[29], 重叠布居数为正, 表示原子之间形成共价键相互作用, 且数值越大共价键越强。Ti12Al11Nb合金延性显著改善也与其内部化学键密不可分。表5给出纯γ-Ti Al, Ti12Al11Nb与Ti11Nb Al12合金中Ti-Ti, Al-Al和Al-Ti的重叠布居数的平均值。三者之间的强度差异程度可由其方差W表示。W为:
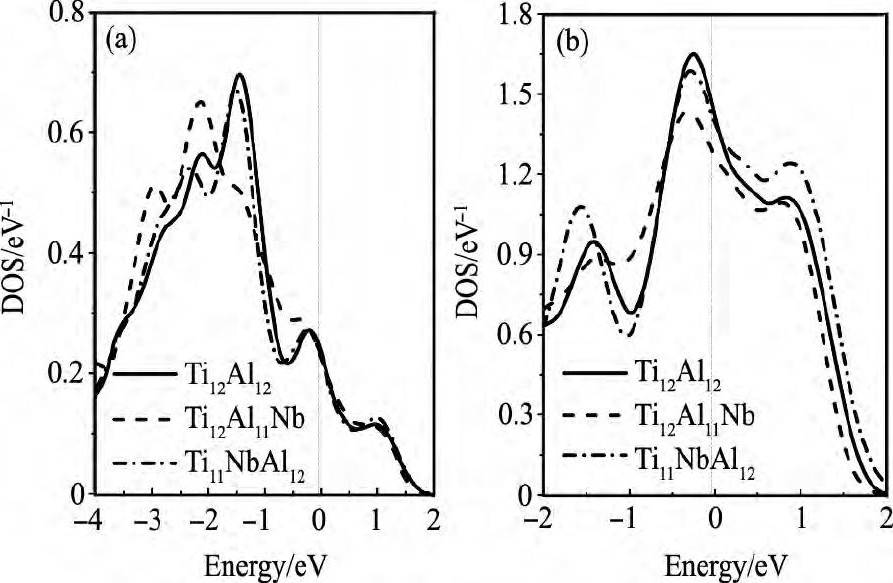
图5 电子态密度Fig.5 DOS
(a) Al-p orbital; (b) Ti-d orbital
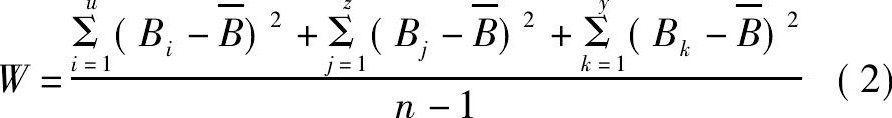
式中, Bi, Bj, Bk分别为Ti-Ti, Al-Al与Al-Ti键重叠布居数的数值;u, z, y分别为Ti-Ti, AlAl与Al-Ti键的个数;  为合金内所有Ti-Ti, Al-Al和Al-Ti键的重叠布居数的平均值;n为Ti-Ti, Al-Al和Al-Ti键的总个数。计算所得的W值也列于表4。
为合金内所有Ti-Ti, Al-Al和Al-Ti键的重叠布居数的平均值;n为Ti-Ti, Al-Al和Al-Ti键的总个数。计算所得的W值也列于表4。
由表4可知, 三合金体系之间共价键的大小关系为:Al-AlTi12Al12>Al-AlTi11Nb Al12>Al-AlTi12Al11Nb;为证明该观点的正确性, 现给出三合金体系同一Al层晶面的电荷密度图, 如图6所示。
为了便于表达, 设D0, D1, D2分别为纯γ-Ti Al, Ti12Al11Nb与Ti11Nb Al12的电荷密度, D0Al-Al表示纯γ-Ti Al中Al与Al原子之间的电荷密度, 其他原子之间同理。对比图6可知, D0Al-Al>D2Al-Al>D1Al-Al, 即, 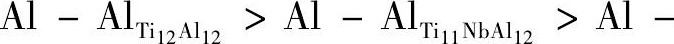
 。该结论与重叠布居数显示结论一致。
。该结论与重叠布居数显示结论一致。
表4 γ-Ti Al基合金体系中的重叠布居数Table 4 Overlap population ofγ-Ti Al based alloys 下载原图
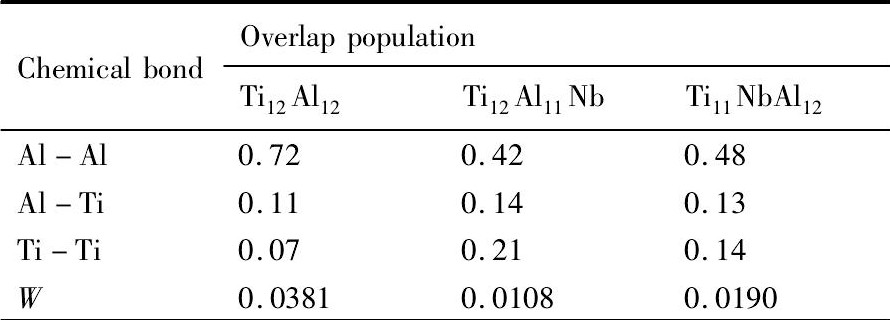
表4 γ-Ti Al基合金体系中的重叠布居数Table 4 Overlap population ofγ-Ti Al based alloys

图6 电荷密度图Fig.6 Charge density maps
(a) Ti12Al12; (b) Ti12Al11Nb; (c) Ti11Nb Al12

上述合金中共价键的变化会导致Al-Al, Al-Ti, Ti-Ti三者之间的强度差异程度发生的变化。由表4可知,  , 这表明Ti12Al11Nb合金中3种主要共价键的强度差异程度小于Ti11Nb Al12与纯γ-Ti Al合金, 即Ti12Al11Nb合金的各向同性程度高于Ti11Nb Al12与纯γ-Ti Al合金。结合结论γ-Ti Al基合金的各向同性程度越高, 该合金的延性越好[22], 即Ti12Al11Nb合金的延性高于Ti11Nb Al12与纯γ-Ti Al合金。
, 这表明Ti12Al11Nb合金中3种主要共价键的强度差异程度小于Ti11Nb Al12与纯γ-Ti Al合金, 即Ti12Al11Nb合金的各向同性程度高于Ti11Nb Al12与纯γ-Ti Al合金。结合结论γ-Ti Al基合金的各向同性程度越高, 该合金的延性越好[22], 即Ti12Al11Nb合金的延性高于Ti11Nb Al12与纯γ-Ti Al合金。
为了解Nb原子与其近邻原子之间的相互作用, 表5给出Ti12Al11Nb与Ti11Nb Al12合金掺杂前后同一位置原子之间的重叠布居数。对于Ti12Al11Nb合金, Nb原子取代Al原子后, Nb-Al键合强度较掺杂前Al-Al键合强度显著降低, 而Nb-Ti之间的重叠布居数为负值, 表明Nb与Ti原子之间没有共价作用;对于Ti11Nb Al12合金, Nb原子取代Ti原子后, Nb-Al与Nb-Ti键均较原来位置的共价键显著减弱。即Nb掺杂使合金的共价性减弱。
表5 Nb与近邻原子之间的重叠布居数Table 5 Overlap population between Nb and its near atoms 下载原图
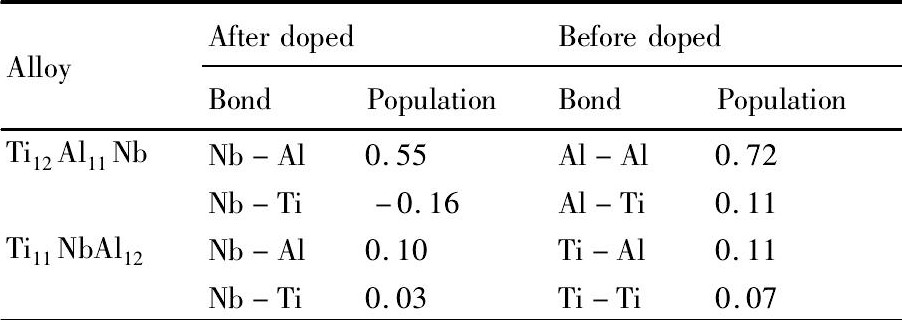
表5 Nb与近邻原子之间的重叠布居数Table 5 Overlap population between Nb and its near atoms
3结论
综合以上对纯γ-Ti Al, Ti1-xNbxAl及Ti Al1-x Nbx (x=1.85%, 2.78%, 4.17%, 6.25%) 系列合金体系的计算与分析, 可以得到以下结论:
1.Nb替位掺杂γ-Ti Al基合金具有较好的能量稳定性, 可以实验制备并能稳定存在。
2.当Nb替位Al掺杂且杂质浓度为1.85%~4.17%时, 能显著改善γ-Ti Al基合金的延性。
3.Nb原子替代Al掺杂使Ti 4s→3d, Al 3s→3p的电荷转移明显减少, p-d杂化作用减弱, 形成金属键的自由电子增多, 金属性增强, 从而使延性得以改善。
4.Nb原子替代Al掺杂会使得合金的共价性减弱, 且Al-Al, Ti-Ti和Al-Ti键之间的强度差异程度明显降低, 这将会显著提高γ-Ti Al基合金的各向同性程度, 从而使该类合金材料的延性得以改善。
参考文献
[1] Castillo-Rodriguez M, No M L, Jimenez J A, Ruano O A, Juan J S.High temperature internal friction in a Ti-46Al-1Mo-0.2Si intermetallic, comparison with creep behaviour[J].Acta Materialia, 2016, 103 (15) :46.
[2] Chen Y Y, Kong F T, Han J C, Chen Z Y, Tian J.Influence of yttrium on microstructure, mechanical properties and deformability of Ti-43Al-9V alloy[J].Intermetallics, 2005, 13 (3-4) :263.
[3] Tao H J, Peng K, Xie Y Q, He Y H, Yin Z M.Research advance on brittleness of Ti-Al intermetallics[J].Materials Science and Engineering of Powder Metallurg, 2007, 12 (6) :330. (陶辉锦, 彭坤, 谢佑卿, 贺跃辉, 尹志民.Ti-Al金属间化合物脆性问题的研究进展[J].粉末冶金材料科学与工程, 2007, 12 (6) :330.)
[4] Zhang J, Feng D, Yin F J.Some new aspects in developing Ti Al based alloys as competitive high temperature materials[J].Advanced Materials, 2011, 278 (9) :557.
[5] Yang R.Advances and challenges of Ti Al base alloys[J].Acta Metallurgica Sinica, 2015, 51 (2) :129. (杨锐.钛铝金属间化合物的进展与挑战[J].金属学报, 2015, 51 (2) :129.)
[6] Hu H, Wu X Z, Wang R, Li W G, Liu Q.Phase stability, mechanical properties and electronic structure of Ti Al alloying with W, Mo, Sc and Yb:first-principles study[J].Journal of Alloys and Compounds, 2016, 658 (15) :689.
[7] Wang H Y, Hu Q K, Yang W P, Li X S.Influence of metal element doping on the mechanical properties of Ti Al alloy[J].Acta Physica Sinica, 2016, 65 (7) :1. (王海燕, 胡前库, 杨文朋, 李旭升.金属元素掺杂对Ti Al合金力学性能的影响[J].物理学报, 2016, 65 (7) :1.)
[8] Song Q G, Zhao J P, Gu W F, Zhen D D, Guo Y R, Li Z P.Ductile and electronic properties of La-doped gamma-Ti Al systems based on density functional theory[J].Acta Physica Sinica, 2017, 66 (6) :191. (宋庆功, 赵俊普, 顾威风, 甄丹丹, 郭艳蕊, 李泽朋.基于密度泛函理论的La掺杂γ-Ti Al体系结构延性与电子性质[J].物理学报, 2017, 66 (6) :191.)
[9] Xia J J, Liang W P, Miao Q, Ren B L, Han P D.First-principles study on electronic structure ofγ-Ti Al alloy doped with Y[J].Rare Metal Materials and Engineering, 2017, 46 (2) :421. (夏金姣, 梁文萍, 缪强, 任蓓蕾, 韩培德.Y掺杂γ-Ti Al电子结构的第一性原理计算[J].稀有金属材料与工程, 2017, 46 (2) :421.)
[10] Shu S L, Qiu F, Tong C Z, Shan X N, Jiang Q C.Effects of Fe, Co and Ni elements on the ductility of Ti Al alloy[J].Journal of Alloys and Compounds, 2014, 617 (15) :302.
[11] Tang S Q, Qu S J, Feng A H, Feng C, Cui K B, Shen J.Recent advances in high temperature oxidation resistance of Ti Al-based alloys[J].Chinese Journal of Rare Metals, 2017, 41 (1) :81. (汤守巧, 曲寿江, 冯艾寒, 冯聪, 崔扣彪, 沈军.Ti Al基合金高温抗氧化研究进展[J].稀有金属, 2017, 41 (1) :81.)
[12] Dong C L, Yu H C, Jiao Z H.Characterization of creep behavior of Ti Al alloy with high Nb content at elevated temperatures[J].Rare Metals, 2016, 35 (1) :106.
[13] Liu Y, Hu R, Kou H C, Wang J, Zhang T B, Li J S, Zhang J.Solidification characteristics of high Nb-containingγ-Ti Al-based alloys with different aluminum contents[J].Rare Metals, 2015, 34 (6) :381.
[14] Fang L, Lin J P, Qiu X C, Qu J X, Ding X F.Turning machining induced microstructural stability of a high Nb-containing Ti Al alloy during high temperature exposure[J].Rare Metals, 2016, 35 (1) :77.
[15] Li H Z, Han B, Hu H T, Zhang X W, Zhu C L, Zhang J.Effect of Nb content on tensile properties of cast Ti Al base alloys with high Al containing at room temperature[J].Chinese Journal of Rare Metals, 2016, 40 (6) :540. (李海昭, 韩波, 胡海涛, 张熹雯, 朱春雷, 张继.Nb含量对铸造高铝Ti Al合金室温拉伸性能的影响[J].稀有金属, 2016, 40 (6) :540.)
[16] Zhang L Z, Wang B Y, Wang D N, Wei L, Lin J P, Wang W J.Study of doping effect of Nb in Ti Al alloy by positron annihilation spectroscopy[J].Acta Metallurgica Sinica, 2007, 43 (3) :269. (张兰芝, 王宝义, 王丹妮, 魏龙, 林均品, 王文俊.正电子湮没谱研究Nb在Ti Al合金中的掺杂效应[J].金属学报, 2007, 43 (3) :269.)
[17] Dang H L, Wang C Y, Yu T.First-principles investigation on alloying effect of Nb and Mo inγ-Ti Al[J].Acta Physica Sinica, 2007, 56 (5) :2838. (党宏丽, 王崇愚, 于涛.γ-Ti Al中Nb和Mo合金化效应的第一性原理研究[J].物理学报, 2007, 56 (5) :2838.)
[18] Wu H L, Zhang W, Gong S K.Effect of Nb on the bonding characteristics of Ti Al intermetallic compounds:a first-principle study[J].Acta Chimica Sinica, 2008, 66 (14) :1669. (吴红丽, 张伟, 宫声凯.Nb元素影响Ti Al金属间化合物键合特征的第一原理计算[J].化学学报, 2008, 66 (14) :1669.)
[19] Morinaga M, Saito J, Yukawa N, Adachi H.Electronic effect on the ductility of alloyed Ti Al compound[J].Acta Metallurgica et Materialia, 1990, 38 (1) :25.
[20] Liu Y L, Li H, Wang S Q, Ye H Q.Nb effects on the structural and mechanical properties of Ti Al alloy:density-functional theory study[J].Journal of Materials Research, 2009, 24 (10) :3165.
[21] Huang B, Duan Y H, Sun Y, Peng M J.Elastic properties and electronic structures of L12-Ti Al3and L12-Ti (Al, Pt) 3:a density functional theory investigation[J].Materials Science Forum, 2015, 817:816.
[22] Eberhart M E, Clougherty D P, Maclaren J M.Charge density topology and its relationship to properties in intermetallic alloys[J].Philosophical Magazine B, 1993, 68 (4) :455.
[23] Paul R, Katherine C E, Philip D F A, Casey F, Malia B W, Aurelio M, Matthias Z, Sorelle A F, Joshua S, Alexander J N.Mchine-learning-assisted materials discovery using failed experiments[J].Nature, 2016, 533 (5) :73.
[24] Song Q G.A preliminary investigation on materials informatics[J].Chinese Science Bulletin, 2004, 49 (2) :210.
[25] Chen G L, Lin J P.Physical Metallurgy Basis of Ordered Intermetallic Compound Structure Materials[M].Beijing:Metallurgical Industry Press, 1999.285. (陈国良, 林均品.有序金属间化合物结构材料物理金属学基础[M].北京:冶金工业出版社, 1999.285.)
[26] Song Q G, Jiang E Y.Study on the structural and energetic properties of two-dimensional ground state of Ag+ion-vacancy in fast ionic conductor AgxTi S2[J].Acta Physica Sinica, 2008, 57 (3) :1823. (宋庆功, 姜恩永.快离子导体AgxTi S2中Ag+离子-空位的二维基态结构与能量性质研究[J].物理学报, 2008, 57 (3) :1823.)
[27] Kawabata T, Tamura T, Izumi O.Effect of Ti/Al ratio and Cr, Nb, and Hf additions on material factors and mechanical properties in Ti Al[J].Metallurgical and Materials Transactions A, 1993, 24 (1) :141.
[28] Huang Y Y, Wu W M, Deng W, Zhong X P, Xiong L Y, Cao M Z, Long Q W.Behavior of Zr and Nb in Ti Al alloy investigated by positron annihilation technique[J].The Chinese Journal of Nonferrous Metals, 2000, 10 (6) :796. (黄宇阳, 吴伟明, 邓文, 钟夏平, 熊良钺, 曹名洲, 龙期威.用正电子湮没技术研究Zr和Nb在Ti Al合金中的行为[J].中国有色金属学报, 2000, 10 (6) :796.)
[29] Liu X K, Liu Y, Zheng Z, Dai J L.Density functional theory study on geometric and electronic structure of Ti and its hydride[J].Rare Metal Materials and Engineering, 2010, 39 (5) :832. (刘显坤, 刘颖, 郑州, 代君龙.Ti及氢化物几何与电子结构的密度泛函理论研究[J].稀有金属材料与工程, 2010, 39 (5) :832.)

