Trans. Nonferrous Met. Soc. China 24(2014) 1853-1858
First principles calculation on ternary stannide phase narrow band gap semiconductor Na2MgSn
Yi-fu WANG1, Qing-lin XIA2, Liu-xian PAN3, Yan YU2
1. College of Mathematics and Computer Science, Key Laboratory of High Performance Computing and Stochastic Information Processing (Ministry of Education), Hunan Normal University, Changsha 410081, China;
2. School of Physics and Electronics, Central South University, Changsha 410083, China;
3. School of Electrical and Information Engineering, Hunan International Economics University, Changsha 410205, China
Received 20 November 2013; accepted 30 April 2014
Abstract: The electronic structures, chemical bonding, elastic and optical properties of the ternary stannide phase Na2MgSn were investigated by using density-functional theory (DFT) within generalized gradient approximation (GGA). The calculated energy band structures show that Na2MgSn is an indirect semiconductor material with a narrow band gap 0.126 eV. The density of state (DOS) and the partial density of state (PDOS) calculations show that the DOS near the Fermi level is mainly from the Na 2p, Mg 3p and Sn 5p states. Population analysis suggests that there are strongly bonded Mg-Sn honeycomb layers in Na2MgSn. Basic physical properties, such as lattice constant, bulk modulus, shear modulus, elastic constants cij were calculated. The elastic modulus E and Poisson ratio ν were also predicted. The results show that Na2MgSn is mechanically stable soft material and behaves in a brittle manner. Detailed analysis of all optical functions reveals that Na2MgSn is a better dielectric material, and reflectivity spectra show that Na2MgSn promise as good coating materials in the energy regions 6.24-10.49 eV.
Key words: stannide phase Na2MgSn; first principles; electronic structures; chemical bonding; elastic properties; optical properties
1 Introduction
In recent decades, a wide variety of alkali-metal stannides have been synthesized and extensively investigated because of their rich structural diversities and novel chemical bonding [1-9]. Their crystal structures exhibit most diverse tin subunits and networks, ranging from 0-D to 3-D. Because of the wide range of valence electron numbers and size between alkali and alkaline-earth metal cations, multiple structural fragments and networks are to be expected in the solid-state structures of the alkali-alkaline-earth-Sn ternary systems. For example, the Li-Ca-Sn, Na-Ca-Sn, Li-Ba-Sn, Na-Ba-Sn and K-Mg-Sn ternary compounds exhibit novel crystal structures with characteristic components, and most of them are semiconductors with energy gaps of 0.11-4.0 eV, and a few of them are metallic [10-13].
Recently, a new hexagonal phase compound Na2MgSn was synthesized [14]. The crystal structure, electronic and physical properties of Na2MgSn, which is built up from 2-D honeycomb layers of [(MgSn)2-] with Na atoms as “space fillers”, were studied by experimental investigation and theoretical calculation [14]. The electrical conductivity experiment and first principles calculation show that Na2MgSn is a narrow band gap semiconductor. However, relatively little is known regarding the mechanical properties, and other optical properties, since these properties are very important for the potential application of Na2MgSn. Motivated by this observation, in this work, a systematical first principles study on the electronic structure, chemical bonding, elastic and optical properties of the hexagonal phase Na2MgSn was done by using first-principles calculations based on DFT.
2 Calculation details
The first principle calculations described here are based on DFT using a plan-wave expansion of the wave function [15,16]. The exchange correlation energy is calculated by the GGA with the Perdew-Burke-Ernzerhof (PBE) function [17]. The ionic cores are represented by ultra-soft pseudopotentials for Na, Mg and Sn atoms. The Na 2s22p63s1 electrons, Mg 3s2 electrons and Sn 5s25p2 electrons are explicitly treated as valence electrons. The Monkhorst and Pack scheme of k-point sampling is used for integration over the first Brillouin zone [18]. The cutoff energy is chosen to be 600 eV, and the Brillouin-zone sampling k-point set mesh parameters are 8×8×6. This set of parameters assure the total energy convergence of 5.0×10-6 eV/atom, the maximum force of 0.01 eV/ 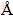 , the maximum stress of 0.02 GPa and the maximum displacement of 5.0×10-4 .
, the maximum stress of 0.02 GPa and the maximum displacement of 5.0×10-4 .
Within the electric-dipole approximation, the imaginary parts of the dielectric functions ε2(ω) can be calculated from the momentum matrix elements between the occupied and unoccupied wave functions within the selection rules, and the real part of dielectric function ε1(ω) can be evaluated from the imaginary part of dielectric function by  relationship [19,20]. Therefore, all solid macroscopical optical constant, such as reflectivity R(ω), energy-loss function L(ω), optical absorption coefficient I(ω), optical conductivity σ(ω), refractive index n(ω) and extinction coefficient k(ω), can be calculated by using the imaginary part of dielectric function ε2(ω) and the real part of dielectric function ε1(ω).
relationship [19,20]. Therefore, all solid macroscopical optical constant, such as reflectivity R(ω), energy-loss function L(ω), optical absorption coefficient I(ω), optical conductivity σ(ω), refractive index n(ω) and extinction coefficient k(ω), can be calculated by using the imaginary part of dielectric function ε2(ω) and the real part of dielectric function ε1(ω).
3 Results and discussion
3.1 Geometry and structure properties
The crystal structure of the hexagonal phase Na2MgSn belongs to the space group P63/mmc (No.194, Z=2), with 2-D honeycomb layers of [(MgSn)2-] and Na atoms as space fillers. There are three inequivalent atomic positions: Na at 4f site (1/3, 2/3, zNa) with zNa=0.5816, Mg at 2b site (0, 0, l/4) and Sn at 2c (l/3, 2/3, l/4) [14]. The lattice parameters are a=5.0486 , c=10.095 .
At the first stage the full structural optimization of this phase was performed both over the lattice parameters and the atomic positions including the internal coordinate. The calculated optimization lattice parameters a, c, V and atomic coordinates compared with available experimental data [14] for Na2MgSn are summarized in Table 1, which shows that the calculated values of GGA calculation are in agreement with the available data.
Table 1 Calculated lattice parameters and atomic internal coordinate compared with available experimental data [14] for Na2MgSn

3.2 Electronic and chemical bonding
The calculated energy band structure of Na2MgSn along with the high-symmetry points of the Brillouin zone by GGA is shown in Fig. 1. The top of the valence band is taken as the zero of energy. This compound is found to have indirect narrow band gap. The valence band maximum (VBM) is at the Γ point and the conduction band minimum (CBM) is at the K point. The calculated band gap value is 0.126 eV by GGA, which is very close to the experimental value of 0.17 eV [14]. The differences between our value and the experimental datum may be due to the well-known underestimation of conduction band state energies in DFT calculations. The total densities of states (TDOS) and partial densities of states (PDOS) for Na2MgSn are plotted in Fig. 2. The DOS near the Fermi level (EF) originates mainly from the Na 2p, Mg 3p and Sn 5p electric states.
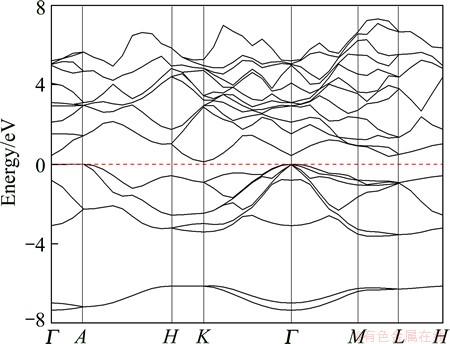
Fig. 1 GGA calculated band structure of Na2MgSn along some high-symmetry lines in Brillouin zone (zero of energy is at the Fermi level)
The Mulliken bond population is useful for evaluating the bonding character in a material. A high value of the bond population indicates a covalent bond, and a low value indicates an ionic bond. Positive and negative values indicate bonding and anti-bonding states, respectively [19,20]. The Mulliken atomic population of Na2MgSn reported in Table 2 shows a significant charge transfer from Na to Mg and Sn, indicating the ionic character between Na and Mg, Sn; the bond population reported in Table 3 shows strong covalent character of Mg-Sn. The chemical bonding in Na2MgSn has predominantly covalent character with mixed covalent- ionic character.
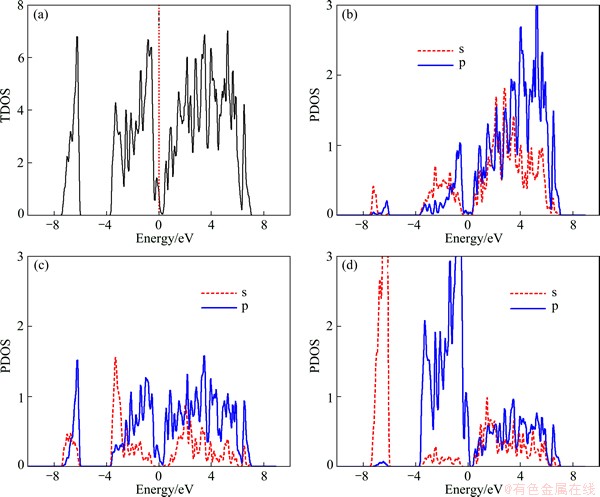
Fig. 2 Total DOS (a) and PDOS of Na (b), Mg (c) and Sn (d) of Na2MgSn calculated by GGA
Table 2 Mulliken atomic population of Na2MgSn
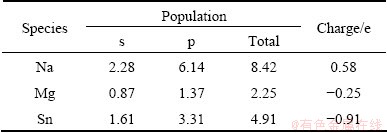
Table 3 Mulliken bond population of Na2MgSn

3.3 Elastic properties
Elastic properties are very important for materials because they provide information on interatomic potentials and relate to various fundamental solid state phenomena such as interatomic bonding, equations of state, phonon spectra as well as specific heat capacity, thermal expansion, Debye temperature [21,22].
Elastic constants are defined by means of a Taylor expansion of the total energy, namely the derivative of the energy as a function of a lattice strain [15,16]. In this work, based on the finite strain technique, the number of steps for each strain is chosen to be 6 and the maximum strain amplitude is 0.002. The hexagonal Na2MgSn crystal has five independent single crystal elastic constants C11, C33, C44, C12 and C13 [23]. The GGA calculated Cij are presented in Table 4. For the hexagonal crystal, its mechanical stability requires that its independent elastic constants should satisfy the Born’s stability criteria [23].
C12>0, C33>0, C11-C12>0, C44>0,
(C11+C12)C33-2 >0 (1)
>0 (1)
Table 4 Calculated single crystal elastic constants Cij, bulk modulus and compressibility coefficient β of Na2MgSn

From Table 4, it can be seen that these criteria are all satisfied, which indicates that Na2MgSn is mechanically stable. The single crystal bulk modulus B is about 24.38 GPa, which indicates that Na2MgSn is a very soft material. The polycrystal bulk modulus B, shear modulus G estimated using the Voigt-Reuss-Hill approach are in the following forms [24-27]:
BH=(BR+BV)/2, GH=(GR+GV)/2 (2)
Elastic modulus E and Poisson ratio ν estimated by
E=9BG/(3B+G),
ν=(3B-E)/6B=(3B-2G)/(6B+2G) (3)
All the calculated results are presented in Table 5. It can be seen that the value of B/G for Na2MgSn is 1.2365, which is smaller than the critical value (1.75) separating ductile and brittle materials [23-28], indicating that Na2MgSn behaves in a brittle manner.
Table 5 Calculated polycrystalline elastic constants, elastic modulus E, Poisson ratio v and B/G of Na2MgSn

It is well known that the values of Poisson ratio (v) are the minimal for covalent materials (ν=0.1), and increase for ionic systems [29]. In our case, the calculated Poisson ratio is 0.1815, which means a sizable ionic contribution in intra-bonding.
The elastic anisotropy in compressibility (AB) and shear modulus (AG) using the model in Ref. [30] for polycrystalline materials is
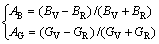 (4)
(4)
A value of zero represents elastic isotropy and a value of 1 is the largest possible anisotropy. The calculated polycrystalline elastic anisotropy in bulk modulus AB is 0.04654 and the shear modulus AG is 0.05680. The shear modulus is somewhat more anisotropy than the bulk modulus.
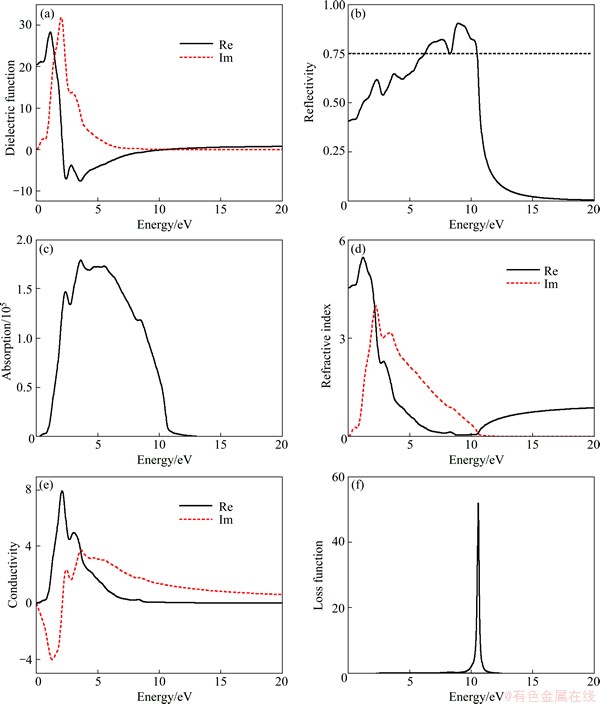
Fig. 3 Real and imaginary parts of dielectric function (a), reflectivity (b), absorption (c), refractive index (d), real and imaginary parts of conductivity (e) and loss function (f) of Na2MgSn
3.4 Optical properties
Figure 3 shows the optical functions of Na2MgSn calculated for photon energies up to 20 eV for polycrystalline sample. The study on the optical functions may help to give a better understanding of the electronic structure. The dielectric function curves as functions of the photon energy are displayed in Fig. 3(a), where the solid line and dashed line represent the real part ε1(ω) and imaginary part ε2(ω), respectively. The calculated static dielectric function value ε1(0) of the Na2MgSn is 20.53. It exhibits metallic characteristics in the energy ranges for ε1(ω)<0. The reflectivity spectra as a function of photon energy are shown in Fig. 3(b). It is found that the reflectivity of Na2MgSn starts with the value of 0.4076, increases and then reaches two maximum values of 0.8203 and 0.9033 at about 7.614 eV and 8.961 eV. The Na2MgSn phase, with reflectivity larger than 0.75 in the energy region 6.24-10.49 eV, may promise as good coating materials. Absorption coefficient is a percentage that tells the decay of light intensity spreads in unit distance in medium. The absorption spectrum shown in Fig. 3(c) reveals the semiconductor nature of Na2MgSn since the spectrum starts from 0.218 eV. According to the relation of refractive index and dielectric function n2-k2=ε1 and 2nk=ε2, the refractive index n and extinction coefficient k of Na2MgSn are obtained. The static refractive index n0 is 4.53, which is in agreement with the calculated result in Fig. 3(a), where the corresponding calculated static dielectric function value ε0(0) is 20.53 ( from 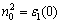 ).
).
Extinction coefficient k indicates the absorption of light and at the same time, it also shows a great absorption characteristic at band-edge. Since the material has no band gap as evident from band structure, the photoconductivity starts with zero photon energy as shown in Fig. 3(e). Loss function L(ω) is an important factor describing the energy loss of a fast electron traversing in a material. The peaks in L(ω) spectrum represent the characteristic associated with the plasma resonance and the corresponding frequency is the so-called plasma frequency. In addition, the peaks of L(ω) also correspond to the trailing edges in the reflection spectra [31,32]. The peak of L(ω) for Na2MgSn presented in Fig. 4(f) is at 10.55 eV, which corresponds to the abrupt reduction of reflectivity spectrum in Fig. 3(b).
To the best of our knowledge, there are no detailed experimental results on the elastic and optical properties of Na2MgSn. We therefore hope that our calculations will motivate experimental studies on the compound.
4 Conclusions
1) The GGA calculated structural parameters of the Na2MgSn are in agreement with the available experimental data. The electronic band structures present that Na2MgSn is a kind of indirect semiconductor material with narrow band gap, and the DOS near the Fermi level is mainly from the p electric states of Na, Mg and Sn.
2) The chemical bonding analysis shows that the Na2MgSn is mainly covalent character with mixed covalent-ionic character. The elastic constants were calculated and the other mechanical parameters were derived. All results show that Na2MgSn is mechanically stable soft material and behaves in a brittle manner.
3) The optical functions of Na2MgSn were calculated and discussed. Reflectivity spectra indicate that the Na2MgSn promises as good coating materials in the energy region of 6.24-10.49 eV.
References
[1] SCHARFE S, KRAUS F, STEGMAIER S, SCHIER A,  T F. Zintl ions, cage compounds, and intermetalloid clusters of group 14 and group 15 elements [J]. Angew Chem Int Ed, 2011, 50: 3630-3670.
T F. Zintl ions, cage compounds, and intermetalloid clusters of group 14 and group 15 elements [J]. Angew Chem Int Ed, 2011, 50: 3630-3670.
[2] IENCO A, HOFFMANN R, PAPOIAN G. Electron-rich bonding and the importance of s, p mixing as one moves across a period: A lesson from the LiSn system [J]. J Am Chem Soc, 2001, 123: 2317-2325.
[3] T F. Structure motifs of sodium stannides on the tin-rich side of the phase diagram [J]. Z Anorg Allg Chem, 2006, 632: 1125-1129.
[4] FORTNER J, SABOUNGI M L, ENDERBY J E. Carrier density enhancement in semiconducting NaSn and CsPb [J]. Phys Rev Lett, 1995, 74: 1415-1418.
[5] HAARMANN F,  D, BEZUGLY V, ROSNER H, GRIN Y. Chemical bonding and solid state NMR of alkali metal monostannides MSn (M=Li, Na, K, Rb, Cs) [J]. Z Anorg Allg Chem, 2006, 632: 1423-1431.
D, BEZUGLY V, ROSNER H, GRIN Y. Chemical bonding and solid state NMR of alkali metal monostannides MSn (M=Li, Na, K, Rb, Cs) [J]. Z Anorg Allg Chem, 2006, 632: 1423-1431.
[6] DUBOIS F, SCHREYER M, T F. NaSn2: A novel binary zintl phase with 2D polyanions of realgar-type units [Sn8]4- [J]. Inorg Chem, 2005, 44(3): 477-479.
[7] PONOU S, KIM S J, T F. Synthesis and characterization of Na5M2+xSn10-x (x≈0.5, M=Zn, Hg)—A doped tetrahedral framework structure [J]. J Am Chem Soc, 2009, 131: 10246-10252.
[8] T F, HOFFMANN S. Na7Sn12: A binary zintl phase with a two-dimensional covalently bonded tin framework [J]. Inorg Chem, 2003, 42(18): 5474-5476.
[9] PONOU S, T F. Nonclassical bonding in the novel structure of Ba2Bi3 and unexpected site preference in the coloring variant Ba2BiSb2 [J]. Inorg Chem, 2004, 43(20): 6124-6126.
[10] TODOROV I, SEVOV S C. Heavy-metal aromatic rings: Cyclopentadienyl anion analogues Sn56- and Pb56- in the zintl phases Na8BaPb6, Na8BaSn6, and Na8EuSn6 [J]. Inorg Chem, 2004, 43(20): 6490-6494.
[11] GE M H, CORBETT J D. M2Ba2Sn6 (M=Yb, Ca): Metallic zintl phases with a novel tin chain substructure [J]. Inorg Chem, 2007 46(10): 4138-4144.
[12] LEI X W. K2Mg5-xSn3 and K3Mg18Tt11 (Tt=Sn, pb) with two types of Mg–Sn/Pb frameworks [J]. J Solid State Chem, 2011, 184: 852-858.
[13] STEGMAIER S, T F. Lithium-stuffed diamond polytype Zn-Tt structures (Tt=Sn, Ge): The two lithium-zinc-tetrelides Li3Zn2Sn4 and Li2ZnGe3 [J]. Inorg Chem, 2013, 52: 2809-2816.
[14] YAMADA T, DERINGER V L, DRONSKOWSKI R, YAMANE H. Synthesis, crystal structure, chemical bonding, and physical properties of the ternary Na-Mg stannide Na2MgSn [J]. Inorg Chem, 2012, 51: 4810-4816.
[15] SEGALL M D, LINDAN PHILIP J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[16] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J, PROBERT M J, REFSON K, PAYNE M C. First principles methods using CASTEP [J]. Zeitschrift fuer Kristallographie, 2005, 220(5-6): 567-570.
[17] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple [J]. Phys Rev Lett, 1996, 77: 3865-3868.
[18] MONKHORST H J, PACK J D. Special points for brillouin-zone integrations [J]. Phys Rev B, 1976, 13: 5188-5192.
[19] HERMET P, GOUMRI-SAID S, KANOUN M B, HENRARD L. First-principles investigation of the physical properties of magnesium nitridoboride [J]. J Phys Chem C, 2009, 113: 4997-5003.
[20] XIA Q L, YI J H, LI Y F, PENG Y D, WANG H Z, ZHOU C S. First-principles investigations of the band structure and optical properties of γ-boron [J]. Solid State Commun, 2010, 150: 605-608.
[21] PONCE C A, CASALI R A, CARAVACA M A. Ab initio study of mechanical and thermo-acoustic properties of tough ceramics: Applications to HfO2 in its cubic and orthorhombic phase [J]. J Phys Condens Mater, 2008, 20: 045213.
[22] BOUHEMADOU A, KHENATA R, CHEGAAR M, MAABED S. First-principles calculations of structural, elastic, electronic and optical properties of the antiperovskite AsNMg3 [J]. Phys Lett A, 2007, 371: 337-343.
[23] SUDHA PRIYANGA G, ASVINI MEENAATCI A T, PALANICHAMY RAJESWARA R, IYAKUTTI K. First principles study of structural, electronic and mechanical properties of transition metal hydrides (TMH, TM=Mo,Tc, Ru) [J]. Transactions of Nonferrous Metals Society of China, 2013, 23: 2700-2707.
[24] SHEIN I R, IVANOVSKII A L. Elastic properties and chemical bonding in ternary arsenide SrFe2As2 and quaternary oxyarsenide LaFeAsO—Basic phases for new 38-55 K superconductors from first principles [J]. Physica C, 2009, 469: 15-19.
[25] SHEIN I R, IVANOVSKII A L. Elastic properties of quaternary oxyarsenide LaOFeAs and LaOFeP as basic phases for new 26-52 K superconducting materials from first principles [J]. Scripta Materialia, 2008, 59: 1099-1102.
[26] SHI Y M, YE S L. Chemical bonding and elastic properties of quaternary arsenide oxides YZnAsO and LaZnAsO investigated by first principles [J]. Transactions of Nonferrous Metals Society of China, 2011, 21: 1378-1382.
[27] HILL R. The elastic behaviour of a crystalline aggregate [J]. Proc Phys Soc London A, 1952, 65: 349-355.
[28] HAINES J, LEGER J M, BOCQUILLON G. Sythesis and design of superhard materials [J]. Rev Mater Res, 2001, 31: 1-23.
[29] ANDERSON O L. A simplified method for calculating the Debye temperature from elastic constants [J]. J Phys Chem Solids, 1963, 24(7): 909-917.
[30] CHUNG D H, BUESSEM W R. Anisotropy in single crystal refractory compounds [M]. Vol. 2. VAHLDIEK F W, MERSOL S A, ed. New York: Plenum Press, 1968: 217.
[31] SUN J, WANG H T, HE J L, TIAN Y J. Ab initio investigations of optical properties of the high-pressure phases of ZnO [J]. Phys Rev B, 2005, 71(12): 125132.
[32] CHENG Y C, WU X L, ZHU J, XU L L, LI S H, CHU PAUL K. Optical properties of rocksalt and zinc blende AlN phases: First-principles calculations [J]. J Appl Phys, 2008, 103: 073707.
三元Stannide相窄帯隙半导体Na2MgSn的第一性原理计算
王一夫1,夏庆林2,潘留仙3,余 燕2
1. 湖南师范大学 数学与计算机科学学院,高性能计算与随机信息处理省部共建教育部重点实验室,长沙 410081;
2. 中南大学 物理与电子学院,长沙 410083;
3. 湖南涉外经济学院 电气与信息工程学院,长沙 410205
摘 要:利用基于密度泛函理论(DFT)的广义梯度近似(GGA)研究Na2MgSn的电子结构、化学键、弹性和光学性质。能带结构显示Na2MgSn为间接带隙材料,带隙宽度为0.126 eV;态密度和分态密度计算结果表明,费米能级附近的态密度主要来自Na、Mg和Sn 的p态电子;布居分析表明Na2MgSn中的化学键具有以共价性为主的混合离子-共价特征。计算得到Na2MgSn的晶格参数、体模量、剪切模量和单晶的弹性常数,由此导出弹性模量和泊松比。结果表明,Na2MgSn是力学稳定的,且具有一定的脆性。光学函数表明Na2MgSn是好的介电材料,反射谱在6.24~10.29 eV的能量范围内,Na2MgSn是较好的被覆材料。
关键词:Stannide 相 Na2MgSn;第一性原理;电子结构;化学键;弹性性质;光学性质
(Edited by Sai-qian YUAN)
Foundation item: Project (11271121) supported by the National Natural Science Foundation of China; Project (11JJ2002) supported by the Natural Science Foundation of Hunan Province, China; Project (11K038) supported by Key Laboratory of High Performance Computing and Stochastic Information Processing of Ministry of Education of China; Projects (2013GK3130, 2014GK3090) supported by the Scientific and Technological Plan of Hunan Province, China
Corresponding author: Qing-lin XIA; Tel: +86-731-88836692, E-mail: qlxia@csu.edu.cn
DOI: 10.1016/S1003-6326(14)63263-5

