өҘЦбАӯУҰБҰПВВБҝХО»РОіЙДЬәНЧФА©ЙўјӨ»оДЬөДјЖЛг
к°ұщ1Ј¬ТЧөӨЗа1, 2Ј¬Бх»¶1Ј¬ҪӯУВ1, 2Ј¬Нхұу1, 2
(1. ЦРДПҙуС§ ІДБПҝЖС§Ул№ӨіМС§ФәЈ¬әюДП іӨЙіЈ¬410083Ј»
2. ЦРДПҙуС§ ҪМУэІҝУРЙ«ҪрКфІДБПҝЖС§Ул№ӨіМЦШөгКөСйКТЈ¬әюДП іӨЙіЈ¬410083)
ХӘТӘЈәФЛУГөЪТ»ФӯАнЖҪГжІЁШНКЖәНNEB(Nudged Elastic Band)№э¶ЙМ¬ЛСЛчөД·Ҫ·ЁјЖЛгУҰБҰЧчУГПВВБөДЧФА©ЙўјӨ»оДЬЈ¬ІўҪбәПFlynnөДФӯЧУЗЁТЖ¶ҜБҰС§АнВЫЈ¬СРҫҝУҰБҰПВВБЧФА©ЙўөДёчПтТмРФЎЈСРҫҝҪб№ыұнГчЈә[100]·ҪПтөДУҰБҰФцҙуВБөДҝХО»РОіЙДЬЈ¬ФЪ6%өДУҰұдПВВБөДҝХО»РОіЙДЬФцҙуБЛ18%ЎЈУҰБҰ¶ФВБЧФА©ЙўјӨ»оДЬөДУ°ПмФЪІ»Н¬·ҪПтҫЯУРІ»Н¬МШХчЈәЛжЧЕУҰБҰөДФцҙуЈ¬ВБөДЧФА©ЙўјӨ»оДЬФЪ[011]·ҪПтЙПјхРЎЈ¬ФЪ[101]·ҪПтЙПФцҙуЎЈУҰБҰ¶ФВБЧФА©ЙўёчПтТмРФөДУ°ПмФЪІ»Н¬јЖЛг·Ҫ·ЁПВУРПаН¬өДЗчКЖЈ¬ө«У°ПміМ¶ИІ»Н¬ЎЈФЪПаН¬УҰБҰПВЈ¬[011]әН[101] 2ёц·ҪПтЧФА©ЙўјӨ»оДЬөДІоТмФЪFlynnДЈРНөДјЖЛгҪб№ыЦРЧоҙуЈ¬ҝјВЗФӯЧУіЪФҘөДFlynnДЈРНөДјЖЛгҪб№ыҙОЦ®Ј¬өЪТ»РФФӯАн№э¶ЙМ¬ЛСЛчөДјЖЛгҪб№ыЧоРЎЎЈ
№ШјьҙКЈәУҰБҰЈ»AlЈ»ЧФА©ЙўјӨ»оДЬЈ»ФӯЧУЗЁТЖ¶ҜБҰС§Ј»өЪТ»ФӯАн№э¶ЙМ¬ЛСЛч
ЦРНј·ЦАаәЕЈәTG111.6 ОДПЧұкЦҫВлЈәA ОДХВұаәЕЈә1672-7207(2013)06-2214-09
Calculation of vacancy formation energy and self-diffusion activation energy of aluminum under uniaxial tensile stress
ZANG Bing1, YI Danqing1, 2, LIU Huan1, JIANG Yong1, 2, WANG Bin1, 2
(1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Key Laboratory of Nonferrous Metal Materials Science and Engineering, Ministry of Education,Central South University, Changsha 410083, China)
Abstract: The self-diffusion activation energy of aluminum under stress was calculated by using the first principle pseudopotential plane wave method and NEB transition state search method. Combined with atomic migration dynamics theory, the anisotropy of the self-diffusion of aluminum under stress was also investigated. The results show that the vacancy formation energy of aluminum increases when the stress is applied, and it increases by 18% under the strain of 6%. The influences of the stress on the self-diffusion activation energy of aluminum are different under different diffusion directions. With the increase of the stress, the self-diffusion activation energy of aluminum decreases in [011] direction, and increases in [101] direction. The influences of the stress on the anisotropy of the self-diffusion calculated with different methods are the same, but the degrees of influence are different. Under the same stress, the ratio of activation energies in [011] and [101] direction is the largest when calculated with FlynnЎҜs atomic migration dynamics theory method smaller with FlynnЎҜs method with atomic relaxation and the smallest with first principle transition state search method.
Key words: stress; Al; self-diffusion activation energy; atomic migration dynamics theory; first principle transition state search method
ФЪҙҝҪрКфәНәПҪрЦРЈ¬А©ЙўУлҫ§ҪзФЛ¶ҜЎўПаұдЎўИдұдөИУРЧЕЗҝБТөДБӘПөЎЈөгИұПЭ(°ьАЁҝХО»әНИЬЦКФӯЧУ)ДЬ№»ФЪИИХЗВдөДЧчУГПВФЪҫ§МеЦРЧчОЮ№жФтА©ЙўЈ¬ТІДЬФЪУҰБҰөДЧчУГПВЧц¶ЁПтөДЗЁТЖЈ¬ІъЙъИЬЦКФӯЧУ(»тҝХО»)А©ЙўөДёчПтТмРФЈ¬ҙУ¶ш¶ФА©ЙўҝШЦЖөДПаұдІъЙъУ°ПмЈ¬ФЪДіР©әПҪрЦРІъЙъЎ°УҰБҰО»ПтР§УҰЎұЎЈОӘБЛҪвКНЎ°УҰБҰО»ПтР§УҰЎұәНСРҫҝЗеіюУҰБҰПВөДФӯЧУА©Йў№эіМЈ¬№ъДЪНвС§ХЯФЪКөСйәНАнВЫЙПҪшРРБЛҙуБҝ№ӨЧч[1-7]ЎЈНхәкО°өИ[4]СРҫҝБЛНвјУУҰБҰ¶Ф2E12ВБәПҪрКұР§РРОӘөДУ°ПмЈ¬ИПОӘНвјУУҰБҰДЬҙЩҪшSПаөДОціцЈ¬ІўІъЙъГчПФөДУҰБҰО»ПтР§УҰЈ¬ІўёщҫЭFlynnөДФӯЧУЗЁТЖ¶ҜБҰС§АнВЫөГіцУҰБҰПВВБөДЧФА©ЙўјӨ»оДЬҫЯУРёчПтТмРФЎЈІЬЛШ·јөИ[5]СРҫҝБЛНвјУУҰБҰ¶ФAl-Cu-Mg-AgәПҪрКұР§РРОӘөДУ°ПмЈ¬·ўПЦФЪНвјУУҰБҰөДУ°ПмПВЈ¬ҰёПаІъЙъУҰБҰО»ПтР§УҰЎЈArdellөИ[6]АыУГФӯЧУЗЁТЖ¶ҜБҰС§АнВЫСРҫҝБЛNi-AlәПҪрФЪУҰБҰПВөДА©ЙўПөКэЈ¬јЖЛгөГөҪ150 MPaөДС№УҰБҰК№ФӯЧУА©ЙўДЬБҰҪөөН6%ЎЈДҝЗ°УҰБҰ¶ФА©ЙўРРОӘУ°ПмөДСРҫҝҙу¶јТФФӯЧУ¶ҜБҰС§АнВЫОӘ»щҙЎЈ¬Г»УРҝјВЗУҰБҰПВөДФӯЧУіЪФҘөИТтЛШЈ¬У°ПмБЛјЖЛгҪб№ыөДҝЙҝҝРФЎЈФЪөЪТ»РФФӯАнЦРТэИлУҰБҰөД·Ҫ·ЁТСҫӯ·ЗіЈіЙКм[7-9]Ј¬ө«ОҙјыУҰБҰПВВБЧФА©ЙўРФЦКөДұЁөАЎЈјшУЪФӯЧУА©Йў¶ҜБҰС§АнВЫөДҫЦПЮРФәНөЪТ»РФФӯАнјЖЛгөДҫ«И·РФЈ¬ұҫОДЧчХЯҙУөЪТ»РФФӯАніц·ўЈ¬јЖЛгіцУҰБҰПВВБөДЧФА©ЙўјӨ»оДЬЈ¬Н¬КұУлФӯЧУЗЁТЖ¶ҜБҰС§АнВЫҪшРРұИҪПЈ¬ёьЧјИ·өДөГөҪУҰБҰ¶ФА©ЙўРРОӘөДУ°ПмЎЈ
1 јЖЛг·Ҫ·Ё
»щУЪГЬ¶И·әәҜАнВЫ(DFT)өДјЖЛг·Ҫ·Ё[10]Ј¬ҙУөЪТ»РФФӯАніц·ўЈ¬Ҫ«ҫ§МеөД¶аөзЧУЧҙМ¬јт»ҜОӘKohn-Sham·ҪіМЈ¬ҪиЦъVASP(Vienna Ab-initio Simulation Package)[11]Инјю°ь, К№УГШНКЖәНЖҪГжІЁ»щЧйЈ¬јЖЛгМеПөөДөзЧУГЬ¶ИЎўЧЬДЬәНҝХО»РОіЙДЬөИЎЈН¬КұІЙУГNEB(Nudged Elastic Band)[12]·Ҫ·ЁСРҫҝөгИұПЭөДЗЁТЖЈ¬С°ХТА©ЙўВ·ҫ¶ЙПөД°°өгЈ¬өГөҪА©ЙўјӨ»оКЖАЭЎЈөЪТ»РФФӯАнјЖЛгҪб№ыөДККУГОВ¶ИКЗ0 KЈ¬0 KТФЙПОВ¶ИПВМеПөөДА©ЙўОКМвРиТӘҪшРРХс¶ҜЖөВКЎўХс¶ҜмШөИОКМвЈ¬ФЪұҫОДЦРІ»ҪшРРҝјВЗЎЈАнПлҫ§МеІЙУГ2ЎБ2ЎБ2өД32ёцВБФӯЧУі¬°ыҪшРРјЖЛгЈ¬Ҫ«АнПлҫ§МеЦРөДТ»ёцВБФӯЧУИҘөфөГИұПЭҫ§МеЎЈА©ЙўКЖАЭјЖЛг№эіМЦРІЙУГөД№э¶ЙМ¬КЗУЙіхКјМ¬әНЧоЦХМ¬өДФӯЧУО»ЦГөИЦөІоЦөөГөҪЎЈЖҪГжІЁәҜКэҪШ¶ПДЬEcutИЎ500 eVЈ»ІјАпФЁЗшІЙУГMonhkorst-PackМШКвkНшёсөг·Ҫ·ЁЈ¬јЖЛгІЙИЎ9ЎБ9ЎБ9өДkНшёсөгЎЈјЖЛгКХБІЙиЦГОӘЈәЧЬМеДЬБҝұд»ҜОӘ9.6ЎБ10-3 kJ/molЈ¬ГҝёцФӯЧУөДКЬБҰРЎУЪ0.1eV/nmЎЈ

Нј1 јЖЛгДЈРНәНУҰБҰ·ҪПт
Fig. 1 Calculation models and stress applied in crystals
ИзНј1ЛщКҫЈ¬ФЪОЮУҰБҰПВЈ¬ҝХО»VПтЖдЦЬО§8ёцҪьБЪФӯЧУөДА©ЙўКЗөИР§өДЈ¬К©јУ[100]·ҪПтөДУҰБҰәуЈ¬УЙУЪУҰБҰ¶Ф(100)ҫ§ГжәН(010)ҫ§ГжјдҫаөДІ»Н¬У°ПмЈ¬А©Йў·ЦОӘ2ЦЦЗйҝцЈәУлУҰБҰ·ҪПтҙ№ЦұөД[011]Ј¬ Ј¬
Ј¬ әН
әН А©Йў·ҪПтәНУлУҰБҰ·ҪПтіЙ45ЎгөД[101]Ј¬
А©Йў·ҪПтәНУлУҰБҰ·ҪПтіЙ45ЎгөД[101]Ј¬ Ј¬
Ј¬ әН
әН А©Йў·ҪПтЎЈОӘБЛСРҫҝ·ҪұгЈ¬ФЪБҪЦЦА©Йў·ҪПтЙП·ЦұрСЎИЎТ»ёц·ҪПтЧчОӘСРҫҝ¶ФПуЈ¬јҙҝХО»ПтAО»ЦГА©ЙўөД[011]·ҪПтәНҝХО»ПтBО»ЦГА©ЙўөД[101]·ҪПтЎЈОӘБЛСРҫҝУҰБҰКЗИзәО¶ФА©ЙўФЪІ»Н¬·ҪПтІъЙъІ»Н¬өДУ°ПмЈ¬ұҫОДҪшРРУҰБҰПВҝХО»РОіЙДЬәНЧФА©ЙўјӨ»оДЬөДјЖЛгЎЈ
А©Йў·ҪПтЎЈОӘБЛСРҫҝ·ҪұгЈ¬ФЪБҪЦЦА©Йў·ҪПтЙП·ЦұрСЎИЎТ»ёц·ҪПтЧчОӘСРҫҝ¶ФПуЈ¬јҙҝХО»ПтAО»ЦГА©ЙўөД[011]·ҪПтәНҝХО»ПтBО»ЦГА©ЙўөД[101]·ҪПтЎЈОӘБЛСРҫҝУҰБҰКЗИзәО¶ФА©ЙўФЪІ»Н¬·ҪПтІъЙъІ»Н¬өДУ°ПмЈ¬ұҫОДҪшРРУҰБҰПВҝХО»РОіЙДЬәНЧФА©ЙўјӨ»оДЬөДјЖЛгЎЈ
ә¬УРөгИұПЭөДҪб№№ФЪИұПЭЦЬО§»бРОіЙұнГжР§УҰЈ¬УГҝйМеІДБПөДјЖЛгІОКэҪшРРјЖЛгҝЙДЬ»бУРОуІоЎЈСРҫҝұнГчК№УГLDAәНGGAШНКЖҪшРРјЖЛгҝЙТФјхРЎұнГжОуІоЈ¬ІўЗТСЎУГLDAШНКЖөДұнГжРЮХэОуІоРЎУЪGGAШНКЖЈ¬№Кұҫ№ӨЧчІЙУГҫЦУтГЬ¶ИҪьЛЖ(LDA)ЎЈ
УҰБҰөДТэИлКЗНЁ№эФЪУҰБҰ·ҪПтЈ¬јҙaЦб·ҪПтЈ¬ТэИлТ»ёцөҘЦбАӯЙмУҰұд[13]
 (1)
(1)
КҪЦРЈәlҰЕОӘУҰұдҰЕКұҫ§°ыaЦб·ҪПтіӨ¶ИЈ»l0ОӘіхКјҫ§°ыФЪaЦб·ҪПтөДіӨ¶ИЎЈН¬КұАыУГөЪТ»РФФӯАн·Ҫ·Ё¶ФФӯЧУҪб№№ҪшРРіЪФҘЈ¬К№ПөНіФЪУҰБҰРОұдПВҙпөҪДЬБҝЧоУЕҪб№№ЎЈЧчУГФЪХыёцПөНіЙПөДУҰБҰҝЙТФёщҫЭNielsen-Martin·Ҫ·Ё»сөГ[14]
 (2)
(2)
ЖдЦРЈәEtОӘМеПөөДЧЬДЬЈ»ҰТОӘУҰБҰЈ»ҰёОӘМеПөөДМе»эЎЈҪ«АнПлВБҫ§МеАӯЙмұдРОјЩЙиОӘөҜРФұдРОЈ¬І»ҝјВЗЛЬРФұдРОЧчУГЈ¬Т»·ҪГжҝЙТФјт»ҜјЖЛгЈ¬БнТ»·ҪГжҝЙТФУлЛыИЛөДөҜРФјЖЛгДЈРНҪшРРұИҪПЎЈNielsen-Martin·Ҫ·ЁКЗК№УГЧЬДЬ¶ФУҰұдЧцЖ«Оў·ЦөД·Ҫ·ЁЗуөГПөНіөДУҰБҰЈ¬ОӘБЛұЈЦӨ5%УҰұдДЪөДҪб№ыөДЧјИ·РФЈ¬УҰұдИЎ0~6%ЎЈФЪјЖЛг№эіМЦРІ»ҝјВЗІҙЛЙР§УҰЈ¬ФЪК©јУУҰБҰөДГҝТ»ІҪ¶јК№УГЙПТ»ІҪУЕ»ҜөДҪб№№ҪшРРіЪФҘЎЈ
2 јЖЛгҪб№ыУл·ЦОц
2.1 І»Н¬УҰұдПВөДУҰБҰ
ёщҫЭNielsen-Martin·Ҫ·ЁЈ¬ОӘБЛөГөҪІ»Н¬АӯУҰұдПВ¶ФУҰөДМеПөЖҪҫщУҰБҰЈ¬КЧПИҪ«МеПөФЪІ»Н¬өДөҘЦбАӯЙмУҰұдПВҪшРРЧЬДЬөДјЖЛгЈ¬өГөҪІ»Н¬УҰұдПВөДЧЬДЬУлОЮУҰұдПВөДЧЬДЬІоЈ¬ФЪХвАпЦ»ҝјВЗБЛФЪөНУҰұдПВөДМеПөРФЦКЈ¬УҰұдИЎ0~6%ЎЈИзНј1ЛщКҫЈ¬ЛжЧЕУҰұдөДФцҙуЈ¬АнПлМеПөөДЧЬДЬЦрҪҘФцҙуЎЈУГөГөҪөДУҰұд-ЧЬДЬ№ШПөҪшРРЖ«Оў·ЦјЖЛгөГөҪІ»Н¬ұдРОМхјюПВ¶ФУҰөДУҰБҰЈ¬ИзНј2ЛщКҫЈ¬ФЪ6%өДУҰұд·¶О§ТФДЪЈ¬ЛжЧЕұдРОіМ¶ИөДФцҙуЈ¬МеПөөДУҰБҰЦрҪҘФцҙуЈ¬ІўЗТіКПЦөҜРФРФЦКЎЈ
ёщҫЭНј2(b)ЦРөДУҰБҰУҰұд№ШПөЈ¬УЙПВКҪ
 (3)
(3)
ҝЙТФөГөҪВБҫ§МеөДөҜРФДЈБҝЈ¬Ey=98 GPaЈ¬ҙуУЪКөСйЦө69 GPa[15]Ј¬ХвКЗУЙУЪLDAөДЎ°№эКшёҝЎұ(over-binding)Р§УҰЈ¬К№УГLDAјЖЛгөДҪб№ыЦРҫ§ёсІОКэЖ«РЎЈ¬өҜРФДЈБҝЖ«ҙуЈ»Н¬КұјЖЛгөДМеПөОӘАнПлҫ§МеЈ¬Г»УРИұПЭЈ¬өјЦВјЖЛгЦөҙуУЪКөСйЦөЎЈФЪјЖЛгҝХО»РОіЙДЬЎўҝХО»ЗЁТЖДЬәНЧФА©ЙўјӨ»оДЬКұЈ¬ХвАп№ШРДөДКЗДЬБҝөДІоТмЈ¬ҙУәуГжөГөҪөДҝХО»РОіЙДЬөИКэЦөУлКөСйЦөөДұИҪПҝЙТФҝҙіцЈ¬ДЬБҝөДІоЦөұИҪПЧјИ·Ј¬ЛщТФК№УГLDAШНКЖКЗәПККөДЎЈ

Нј2 І»Н¬УҰұдПВөДМеПөЧЬДЬІоәНУҰБҰ
Fig. 2 Total energy difference under different strains and strain-stress curve
2.2 УҰБҰПВөДҝХО»РОіЙДЬЎўА©ЙўКЖАЭәНЧФА©ЙўјӨ»оДЬ
ә¬УРөгИұПЭөДҪб№№Ј¬УлИұПЭҪьБЪөДФӯЧУҙжФЪТ»ёцХжҝХІгОӘФӯЧУјдҫаөДОўРЎұнГжЈ¬ҪшРРЧЬДЬјЖЛгКұ»бҪ«ХвёцұнГжөДДЬБҝ°ьә¬ФЪДЪЈ¬ФЪИұПЭЦЬО§»бРОіЙЎ°ұнГжР§УҰЎұЈ¬УГҝйМеІДБПөДјЖЛгІОКэҪшРРјЖЛг»бУРОуІоЈ¬ФЪјЖЛгҝХО»РОіЙДЬәНЗЁТЖДЬөД№эіМЦРТ»°гТӘҪшРРұнГжРЮХэЎЈСРҫҝұнГчК№УГLDAәНGGAШНКЖҪшРРјЖЛгҝЙТФјхРЎұнГжОуІоЎЈ¶ФУЪВБҫ§МеАҙЛөЈ¬К№УГLDA(GGA)ШНКЖјЖЛгҝХО»РОіЙДЬөДұнГжРЮХэЦөОӘ0.06 eV(0.15 eV)Ј¬јЖЛгЗЁТЖДЬөДұнГжРЮХэЦөОӘ0.02 eV(0.05 eV)[16-17]ЎЈУЙУЪLDAөДұнГжРЮХэЦөәЬРЎЈ¬ІўЗТ·ўПЦјЖЛгөГөҪөДҝХО»РОіЙДЬәНЗЁТЖДЬУлКөСйЦөОуІоәЬРЎЈ¬ЛщТФХвАпГ»УРҝјВЗұнГжРЮХэЎЈ
ҝХО»РОіЙДЬөДјЖЛ㹫КҪОӘ
 (4)
(4)
КҪЦРЈәEd(ҰЕ)ОӘә¬УРТ»ёцҝХО»өДМеПөөДЧЬДЬЈ»Ep(ҰЕ)ОӘАнПлМеПөөДЧЬДЬЈ»NОӘАнПлҫ§МеөДAlФӯЧУёцКэЈ¬АнПлМеПөәНИұПЭМеПө¶јҫӯ№эБЛід·ЦіЪФҘЎЈјЖЛгҪб№ыұнГчЈәГ»УРУҰБҰөДМхјюПВЈ¬ВБөДҝХО»РОіЙДЬОӘ0.695 eVЈ¬УлКөСйЦө0.67 eV[18]»щұҫОЗәПЎЈИзұн1ЛщКҫЈ¬өұМеПөК©јУУҰБҰКұЈ¬ВБөДҝХО»РОіЙДЬФцҙуЈ¬ІўЗТЛжЧЕУҰБҰөДФцјУЈ¬ҝХО»РОіЙДЬЦрҪҘФцҙуЈ¬ФЪ6%өДУҰұдМхјюПВЈ¬ВБөДҝХО»РОіЙДЬОӘ0.826 eVЈ¬УлІ»К©јУУҰБҰПаұИФцҙуБЛҙуФј18%ЎЈ
ФЪ[100]·ҪПтөДөҘЦбУҰБҰПВЈ¬АнПлМеПөәНә¬ҝХО»МеПөөДЧЬДЬ¶јЛжЧЕУҰБҰөДФцҙу¶шФцҙуЈ¬УЙУЪЖдФцјУіМ¶ИІ»Н¬өјЦВҝХО»РОіЙДЬІ»Н¬Ј¬№КУГУҰБҰПВМеПөөДЧЬДЬУлОЮУҰБҰПВМеПөөДЧЬДЬІоұнКҫЖдФцјУіМ¶ИЈә
 (5)
(5)
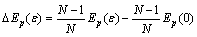 (6)
(6)
КҪЦРЈәЎчEd(ҰЕ) ОӘУҰБҰПВИұПЭМеПөЧЬДЬөДФцјУЈ»ЎчEp(ҰЕ)ОӘУҰБҰПВАнПлПөНіЧЬДЬөДФцјУЎЈИзНј3ЛщКҫЈ¬АнПлМеПөәНә¬ҝХО»МеПөФЪУҰБҰПВУл·ЗУҰБҰПВЧЬДЬІо¶јЛжЧЕУҰБҰөДФцҙу¶шФцҙ󣬶шАнПлМеПөөДЧЬДЬІоФцјУөДіМ¶ИҪПРЎЎЈУЙУЪАнПлМеПөФЪУҰБҰөДЧчУГПВФӯЧУҫщҙҰУЪөгХуёсөгО»ЦГЈ¬МеПөөДЧЬДЬұд»ҜРЎЈ¬¶шК©јУУҰБҰөДИұПЭМеПөҝХО»ЦЬО§өДФӯЧУФЪіЪФҘөДЧчУГПВЈ¬ФӯЧУПтҝХО»ёҪҪьТЖ¶ҜЈ¬Ж«АлөгХуёсөгО»ЦГЈ¬ҪшТ»ІҪФцҙуБЛФӯЧУјдҫаЈ¬МеПөЧЬДЬұд»ҜПа¶Фҙ󣬶шХвҫНөјЦВБЛҝХО»РОіЙДЬЛжЧЕУҰБҰөДФцҙу¶шФцҙуЎЈ

Нј3 УҰБҰПВМеПөөДЧЬДЬУлОЮУҰБҰПВМеПөөДЧЬДЬІоЛжУҰБҰөДұд»Ҝ
Fig. 3 Total energy difference of configuration with stress and without stress as function of strain
К№УГөЪТ»ФӯАнNEB№э¶ЙМ¬ЛСЛчөД·Ҫ·ЁјЖЛгөДҝХО»ЗЁТЖДЬҪб№ыИзНј4ЛщКҫЈ¬ОЮУҰБҰМхјюПВВБөДҝХО»ЗЁТЖДЬјЖЛгҪб№ыОӘ0.608 eVЈ¬КөСйЦөОӘ0.61 eV[18]ЎЈУЙНј4ҝЙЦӘЈәФЪУлУҰБҰ·ҪПтҙ№ЦұөД[011]А©ЙўВ·ҫ¶әНУлУҰБҰ·ҪПтІ»ҙ№ЦұөД[101]А©ЙўВ·ҫ¶ЙПЈ¬УҰБҰ¶јК№ЗЁТЖДЬјхРЎЈ¬ө«јхРЎөДіМ¶ИІ»Т»СщЎЈФЪ[011]А©Йў·ҪПтЙПЈ¬УҰБҰҙу·щ¶ИөДјхРЎБЛҝХО»А©ЙўЗЁТЖДЬЈ¬Изұн1ЛщКҫЈ¬6%өДУҰұдК№ЗЁТЖДЬҪөөНөҪ0.408 eVЈ¬ПВҪөБЛ33%Ј»ФЪ[101]А©Йў·ҪПтЙПЈ¬6%өДУҰұдПВЗЁТЖДЬОӘ0.545 eVЈ¬ПВҪөБЛ10%ЎЈ
[100]·ҪПтөДУҰБҰФцҙуБЛВБөДҝХО»РОіЙДЬЈ¬¶шјхРЎБЛВБФЪ[011]әН[101]А©Йў·ҪПтЙПөДҝХО»ЗЁТЖДЬЈ¬УҰБҰ¶ФЧФА©ЙўјӨ»оДЬөДУ°ПмКЗ¶ФҝХО»РОіЙДЬәНҝХО»ЗЁТЖДЬөДЧЫәПУ°ПмЈ¬ЧФА©ЙўјӨ»оДЬјЖЛ㹫КҪОӘЈә
 (7)
(7)
КҪЦРЈәQОӘЧФА©ЙўјӨ»оДЬЈ»ЎчHfОӘҝХО»РОіЙДЬЈ»ЎчHmОӘҝХО»ЗЁТЖДЬЎЈ
УЙұн1өДКэҫЭҝЙЦӘЈәОЮУҰБҰЧҙМ¬ПВВБөДЧФА©ЙўјӨ»оДЬОӘ1.303 eVЈ¬УлКөСйЦө1.31 eV[18]ОЗәПЎЈУҰұд¶ФІ»Н¬·ҪПтЙПөДЧФА©ЙўјӨ»оДЬІъЙъБЛЦКөДІоТмЈәЛжЧЕУҰБҰөДФцҙуЈ¬[101]·ҪПтЙПЧФА©ЙўјӨ»оДЬФцҙуЈ¬[011]·ҪПтЙПөДЧФА©ЙўјӨ»оДЬјхРЎЈ¬МеПЦіцБЛУҰБҰ¶ФА©ЙўІъЙъөДёчПтТмРФЎЈ

Нј4 І»Н¬УҰБҰПВВБФЪ[011]әН[101]·ҪПтЙПөДЧФА©ЙўДЬАЭ
Fig. 4 Self-diffusion barriers of aluminum in different diffusion directions
ұн1 УҰБҰПВҝХО»РОіЙДЬЎўЗЁТЖДЬәНЧФА©ЙўјӨ»оДЬ
Table 1 Vacancy formation energy, migration energy and self-diffusion activation energy of aluminum under stress

2.3 өзәЙГЬ¶И
ОӘБЛЦұ№ЫөШАнҪвФӯЧУјдөДјьәПЧчУГРФЦКЈ¬»жЦЖБЛІ»Н¬МеПөөДөзәЙГЬ¶И·ЦІјЗйҝцЎЈНј5ЛщКҫОӘАнПлҫ§МеөДөзЧУГЬ¶ИНјЎЈұИҪПНј5(a)әННј5(b)ҝЙЦӘЈ¬УлОЮУҰБҰЧчУГПаұИЈ¬УҰБҰЧчУГПВУлУҰБҰ·ҪПтҙ№ЦұөДAl(100)ГжөДөзЧУГЬ¶И»щұҫІ»ұдЈ¬ІўЗТВБФӯЧУЦЬО§өДөзәЙГЬ¶ИКЗ¶ФіЖөДЎЈУҰБҰПВAl(010)ГжөДөзЧУГЬ¶Иұд»ҜҪПҙуЈ¬ИзНј5(d)ЛщКҫЈ¬УЙУЪЖҪРРУЪҙЛГжөДУҰБҰөДЧчУГЈ¬[100]·ҪПтөДВБФӯЧУјдҫаФцҙуЈ¬AlЎӘAlјьөДЧчУГјхИхЈ¬өзЧУГЬ¶ИјхРЎЈ¬Н¬КұФӯЧУјдҫаөД·З¶ФіЖұд»ҜФміЙВБЦЬО§өзЧУГЬ¶ИөД·З¶ФіЖРФЎЈОӘБЛҪ«І»Н¬А©Йў·ҪПтөДФӯЧУөДөзЧУГЬ¶ИҪшРРұИҪПЈ¬ЧцН¬Кұә¬УРAәНBО»ЦГөД ГжөДөзЧУГЬ¶ИНјЈ¬ИзНј5(c)әННј5(d)ЛщКҫЈ¬ОЮУҰБҰЧчУГПВЈ¬AlЎӘAlјьөДөзЧУГЬ¶ИОӘ32.8 e/nm3Ј¬УҰБҰК№Фӯұҫ¶ФіЖөДөзЧУГЬ¶И·ўЙъ·ҪПтРФұд»ҜЈ¬ФЪУҰБҰЧчУГПВЈ¬[011]·ҪПтөДIЗшУтөДөзЧУГЬ¶ИОӘ32.6 e/nm3Ј¬УлОЮУҰБҰЧчУГөДөзЧУГЬ¶И»щұҫПаөИЈ¬[101]·ҪПтөДIIIЗшУтөДөзЧУГЬ¶ИәН[110]·ҪПтөДIIЗшУтөДөзЧУГЬ¶ИОӘ30.9 e/nm3Ј¬УлОЮУҰБҰЧҙМ¬ПаұИјхРЎБЛФј6%Ј¬ХвКЗТтОӘУҰБҰ¶ФІ»Н¬·ҪПтөДФӯЧУҫаАлІъЙъБЛУ°ПмЎЈ
ГжөДөзЧУГЬ¶ИНјЈ¬ИзНј5(c)әННј5(d)ЛщКҫЈ¬ОЮУҰБҰЧчУГПВЈ¬AlЎӘAlјьөДөзЧУГЬ¶ИОӘ32.8 e/nm3Ј¬УҰБҰК№Фӯұҫ¶ФіЖөДөзЧУГЬ¶И·ўЙъ·ҪПтРФұд»ҜЈ¬ФЪУҰБҰЧчУГПВЈ¬[011]·ҪПтөДIЗшУтөДөзЧУГЬ¶ИОӘ32.6 e/nm3Ј¬УлОЮУҰБҰЧчУГөДөзЧУГЬ¶И»щұҫПаөИЈ¬[101]·ҪПтөДIIIЗшУтөДөзЧУГЬ¶ИәН[110]·ҪПтөДIIЗшУтөДөзЧУГЬ¶ИОӘ30.9 e/nm3Ј¬УлОЮУҰБҰЧҙМ¬ПаұИјхРЎБЛФј6%Ј¬ХвКЗТтОӘУҰБҰ¶ФІ»Н¬·ҪПтөДФӯЧУҫаАлІъЙъБЛУ°ПмЎЈ
Нј6ЛщКҫОӘАнПлҫ§МеФЪУҰБҰЧчУГПВБЩҪьВБФӯЧУҫаАлөДұд»ҜКҫТвНјЎЈУЙНј6ҝЙјыЈәУлУҰБҰ·ҪПтҙ№ЦұөДҫ§ГжЙПөДВБФӯЧУјдҫа»щұҫІ»ұдЈ¬УлУҰБҰ·ҪПтІ»ҙ№ЦұөДҫ§ГжЙПөДВБФӯЧУјдҫаұдҙуЈ¬УЙҙЛөјЦВБЛөзЧУГЬ¶ИөДПаУҰұд»ҜЎЈ
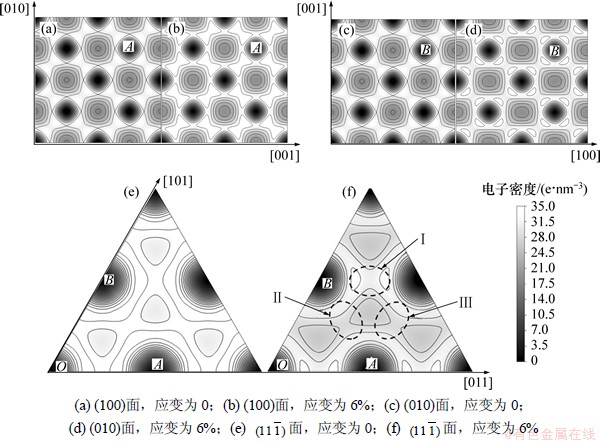
Нј5 АнПлҫ§МеФЪІ»Н¬ГжөДөзЧУГЬ¶И
Fig. 5 Electron density of perfect crystal in different crystal planes

Нј6 АнПлҫ§МеУҰБҰПВВБҪьБЪФӯЧУҫаАлұд»ҜКҫТвНј
Fig. 6 Schematic of change in distance between the nearest neighbors of perfect system under stress
Нј7ЛщКҫОӘә¬ҝХО»ҫ§МеФЪІ»Н¬ГжөДөзЧУГЬ¶ИНјЈ¬УҰБҰ¶Фә¬ҝХО»МеПөөзЧУГЬ¶ИөДУ°ПмУл¶ФАнПлМеПөөДУ°ПмПаЛЖЈ¬ҙУНј7(a)әННј7(b)өДAl(100)ГжөДөзЧУГЬ¶ИҝЙТФҝҙіцЈәУлОЮУҰБҰЧҙМ¬ПаұИЈ¬УҰБҰПВҝХО»ЦЬО§ҪьБЪВБФӯЧУЦ®јдөДөзЧУГЬ¶И»щұҫІ»ұдЈ¬ОӘ32.8 e/nm3Ј¬¶ш(010)Гж(Нј7(c)әННј7(d))өДҝХО»ЦЬО§ҪьБЪВБФӯЧУЦ®јдөДөзЧУГЬ¶ИФтұд»ҜҪПҙуЈ¬ҙУ32.8 e/nm3јхРЎөҪ31.0 e/nm3ЎЈНј7(e)әННј7(f)ЛщКҫОӘН¬Кұ°ьә¬ҝХО»ЎўAО»ЦГәНBО»ЦГөДГжЈ¬ОЮУҰБҰЧҙМ¬ПВЈ¬ҝХО»ЦЬО§6ёцВБФӯЧУјдөДөзЧУГЬ¶ИКЗПаН¬өДЈ¬УҰБҰЧҙМ¬ПВЈ¬[011]·ҪПтAО»ЦГөДВБФӯЧУБҪІаөДөзЧУГЬ¶ИПаН¬Ј¬¶ш[101]·ҪПтBО»ЦГөДВБФӯЧУБҪІаөДөзЧУГЬ¶ИФтІ»Н¬ЎЈ
УҰБҰ¶ФҝХО»ЦЬО§ВБФӯЧУҫаАлөДУ°ПмИзНј8ЛщКҫЈ¬УҰБҰЧҙМ¬ПВЈ¬НкИ«іЪФҘөД[011]·ҪПтөДAО»ЦГВБФӯЧУУлҝХО»өДҫаАлјхРЎЈ¬ө«ЖдУлҝХО»ЦЬО§ЖдЛыО»ЦГ(BЈ¬C)өДВБФӯЧУөДҫаАл¶јФцҙуЈ¬ІъЙъНј(f)ЦРөДAБҪІаөзЧУГЬ¶ИөД¶ФіЖРФЎЈ[101]·ҪПтөДBО»ЦГВБФӯЧУУлҝХО»өДҫаАлФцҙуЈ¬УлAО»ЦГВБФӯЧУөДҫаАлФцҙуЈ¬ө«УлCО»ЦГВБФӯЧУөДҫаАлјхРЎЈ¬ХвЦЦФӯЧУҫаАлөДІ»Н¬ФміЙБЛНј7(f)ЦРөДөзЧУГЬ¶ИөД·З¶ФіЖРФЎЈҝХО»УлAәНBО»ЦГВБФӯЧУөДҫаАлјҙА©Йў№эіМЦРөДФҫЗЁКЖАЭЈ¬УҰБҰ¶ФІ»Н¬·ҪПтФҫЗЁКЖАЭөДУ°ПмөјЦВБЛІ»Н¬·ҪПтЙПЧФА©ЙўјӨ»оДЬөДЗшұрЎЈ
3 МЦВЫ
УҰБҰУлФӯЧУөДА©ЙўјӨ»оДЬөД№ШПөУлҝХО»УлҪьБЪФӯЧУЎўҪьБЪФӯЧУЦ®јдөДҫаАлУРЧЕГЬЗР№ШПөЈ¬ҙУ20КАјН60ДкҙъТФАҙЈ¬Па№ШС§ХЯҫНХвёцОКМвҪшРРБЛСРҫҝЈ¬ЖдЦРЧоОӘЦшГыөДКЗFlynnөДФӯЧУЗЁТЖБҰС§АнВЫ[19]Ј¬ёГАнВЫК№УГөҜРФіЈКэәНУҰБҰПВөДФҫЗЁҫаАлЎўА©ЙўКЖАЭЧчОӘКдИлұдБҝЈ¬Ҫ«УҰБҰУлА©ЙўөД№ШПөұнКҫіцАҙЎЈөұКұФӯЧУЗЁТЖБҰС§АнВЫЦ»КЗҝјВЗУҰБҰ¶ФФӯЧУјдҫаөДУ°ПмЈ¬ІўГ»ДЬҝјВЗУҰБҰПВөДФӯЧУіЪФҘЎЈФЪјЖЛг»ъСёЛЩ·ўХ№өДҪсМмЈ¬К№УГөЪТ»РФФӯАнКЦ¶ОҪшРРУҰБҰПВөДФӯЧУіЪФҘТСҫӯәЬИЭТЧЈ¬ЛщТФҪ«ФӯЧУіЪФҘәуөДФҫЗЁҫаАләНА©ЙўКЖАЭЧчОӘФӯЧУЗЁТЖ¶ҜБҰС§АнВЫөДКдИлІОКэҪшРРјЖЛгЈ¬»бФЪТ»¶ЁіМ¶ИЙПК№Ҫб№ыҫ«И·ЎЈК№УГNEB№э¶ЙМ¬ЛСЛчөД·Ҫ·ЁјЖЛгА©ЙўјӨ»оДЬІ»РиТӘЗЁТЖ¶ҜБҰС§АнВЫДЈРНЦРЛщРиТӘөДБҰС§іЈКэөИКөСйКэҫЭЈ¬НкИ«ҙУөЪТ»РФФӯАніц·ўЈ¬Ҫб№ы»бёьЧјИ·ЎЈ

Нј7 ә¬ҝХО»ҫ§МеФЪІ»Н¬ГжөДөзЧУГЬ¶И
Fig. 7 Electron density of defect crystal in different crystal planes

Нј8 ИұПЭМеПөУҰБҰПВҝХО»УлҪьБЪВБФӯЧУҫаАлұд»ҜКҫТвНј
Fig. 8 Schematic of change in distance between vacancy and the nearest neighbors of defect system under stress
3.1 ФӯЧУЗЁТЖ¶ҜБҰС§АнВЫДЈРН(FlynnЎҜs model)
өұ¶ФТ»Бў·Ҫҫ§МеК©јУНвјУөҘПтАӯЙмУҰБҰ(ҰТ1=ҰТ)КұЈ¬Бў·Ҫҫ§МеҪ«»бСШНвБҰ·ҪПт·ўЙъТ»ёцК®·ЦОўРЎөДөҜРФұдРОЈ¬ФӯЧУјдөДПа¶ФҫаАл·ўЙъұд»ҜЈ¬К№өГФҫЗЁҫаАлl(ҝХО»УлЖдҪьБЪФӯЧУөДҫаАл)әНКЖАЭҫаАлy(ҝХО»ЦЬО§өДҙОҪьБЪФӯЧУҫаАл)·ўЙъұд»ҜЈ¬Хв»б¶ФА©Йў№эіМІъЙъУ°ПмЎЈёщҫЭFlynnөДФӯЧУЗЁТЖ¶ҜБҰС§АнВЫЈ¬ФӯЧУА©ЙўјӨ»оДЬQУлөҜРФіЈКэCәНөҜРФУҰұдҰГЦ®јдөД№ШПөҝЙТФұнКҫОӘЈә
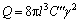 (8)
(8)
ёщҫЭОуІоАнВЫЈ¬ҝЙТФҪ«ЙПКцұнҙпКҪЦРёчёцІОКэЦ®јдөДұд»Ҝ№ШПөұнКҫОӘЈә
 (9)
(9)
Ҫ«ЎчҰГ/ҰГЧӘ»»іЙ-Ўчy/yЈ¬№ККҪ(9)ҝЙұнКҫОӘЈә
 (10)
(10)
АыУГөҜРФБҰС§әНјёәО№ШПөЈ¬ІўЗТәцВФ¶юҪЧРЎПоҰЕ2Ј¬ҝЙТФөГөҪЈә
 Ј¬
Ј¬
 (11)
(11)
КҪЦРЈә Ј»
Ј» ЎЈ
ЎЈ
А©ЙўјӨ»оДЬөДұд»ҜУлФҫЗЁҫаАлlәНКЖАЭҫаАлyУР№ШЈ¬¶шХвБҪХЯФЪ[100]·ҪПтөДНвБҰЧчУГПВ·ЦұрУР2ёцІ»Н¬өДЦөЈ¬ОӘl[011]Ј¬l[101]Ј¬y[011]әНy[011]Ј¬УЙКҪ(10)ҝЙТФ·ЦұрјЖЛгБҪХЯөДұд»ҜөГөҪјӨ»оДЬөДұд»ҜЎЈ
3.2 ҝјВЗФӯЧУіЪФҘөДFlynnДЈРН
Ц®З°УРС§ХЯАыУГFlynnАнВЫҪшРРСРҫҝКұЈ¬КЗАыУГөҜРФіЈКэјЖЛгөГөҪІ»Н¬УҰұдПВөДФҫЗЁҫаАләНКЖАЭҫаАлөДұд»ҜЈ¬ҙУ¶шјЖЛгіцІ»Н¬УҰұдәНУҰБҰПВөДА©ЙўјӨ»оДЬЈ¬ІўГ»УРҝјВЗҝХО»ЦЬО§ФӯЧУөДіЪФҘЎЈХл¶ФХвТ»ОКМвЈ¬ұҫОДМЦВЫБЛіЪФҘ¶ФФҫЗЁҫаАләНКЖАЭҫаАлөДУ°ПмЈ¬ІўАыУГөЪТ»ФӯАніЪФҘЦ®әуөДФӯЧУО»ЦГөГөҪФҫЗЁҫаАләНКЖАЭҫаАлЈ¬ФЪFlynnАнВЫөД»щҙЎЙПјЖЛгБЛІ»Н¬·ҪПтЙПА©ЙўјӨ»оДЬөДұИЦөЎЈ
ОӘБЛөГөҪіЪФҘ¶ФФҫЗЁҫаАлlәНКЖАЭҫаАлyөДУ°ПмЈ¬ЧчОҙіЪФҘәНіЪФҘәуөДlәНyЛжНвБҰөДұд»Ҝ№ШПөНјЈ¬ИзНј9ЛщКҫЎЈҙУНј9ҝЙТФҝҙіцЈәіЪФҘ¶ФlәНyөДКэЦөТФј°ЛжУҰБҰөДұд»ҜРұВК¶јУРУ°ПмЎЈОҙіЪФҘөДФӯЧУЛжЧЕУҰБҰөДФцҙуЈ¬ФЪ[011]·ҪПтөДФҫЗЁҫаАлФцҙуЈ¬КЖАЭҫаАлјхРЎЈ¬ФЪ[101]·ҪПтөДФҫЗЁҫаАлјхРЎЈ¬КЖАЭҫаАлФцҙуЎЈіЪФҘөДҪб№ыЦР[011]әН[101]2ёц·ҪПтЙПөДКЖАЭҫаАл¶јЦрҪҘФцҙуЈ¬ө«ФҫЗЁҫаАлlФтМеПЦіцІ»Н¬өДМШХчЈә[011]А©Йў·ҪПтЙПөДФҫЗЁҫаАлјхРЎЈ¬[101]А©Йў·ҪПтЙПөДФҫЗЁҫаАлФцҙуЎЈУЙКҪ(10)ҝЙЦӘЈәКЖАЭҫаАлyЛжУҰБҰұд»ҜөДРұВКәНФҫЗЁҫаАлlЛжУҰБҰұд»ҜөДРұВК№ІН¬У°ПмЧЕЧФА©ЙўјӨ»оДЬөДұд»ҜЎЈ
Нј10ЛщКҫОӘНвБҰЧчУГПВУлОЮНвБҰПВЧФА©ЙўјӨ»оДЬөДұИЦөФЪІ»Н¬А©Йў·ҪПтЙПөДЛжУҰБҰөДұд»ҜЎЈҙУНјЦРҝЙТФҝҙіцЈ¬ЧФА©ЙўјӨ»оДЬФЪ[011]·ҪПтЙПЛжУҰБҰөДФцҙу¶шјхРЎЈ¬ФЪ[101]·ҪПтЙПЛжУҰБҰөДФцҙу¶шФцҙуЎЈФЪПаН¬өДУҰБҰМхјюПВЈ¬УҰБҰ¶Ф[011]·ҪПтЧФА©ЙўјӨ»оДЬөДУ°ПмТӘРЎУЪ¶Ф[101]·ҪПтЧФА©ЙўјӨ»оДЬөДУ°ПмЎЈ¶ФұИFlynnДЈРНәНҝјВЗФӯЧУіЪФҘөДFlynnДЈРНҝЙТФҝҙіцЈәФӯЧУіЪФҘК№УҰБҰ¶ФА©ЙўРРОӘөДУ°ПмјхРЎЈ¬ХвөгФЪ[011]А©Йў·ҪПтЙПУИОӘГчПФЎЈ
3.3 өЪТ»РФФӯАнУлФӯЧУЗЁТЖ¶ҜБҰС§Ҫб№ыұИҪП
НЁ№эІ»Н¬·Ҫ·ЁјЖЛгөГөҪөДЧФА©ЙўјӨ»оДЬФЪІ»Н¬А©Йў·ҪПтЙПҫщМеПЦіцБЛІоТмРФЈ¬К№УГЧФА©ЙўјӨ»оДЬФЪ[011]А©Йў·ҪПтәН[101]А©Йў·ҪПтЙПөДұИЦөұнХчА©ЙўөДёчПтТмРФЈ¬ИзНј11ЛщКҫЎЈУЙНј11ҝЙјыЈәЛжЧЕУҰБҰөДФцҙуЈ¬2ёц·ҪПтЧФА©ЙўјӨ»оДЬөДұИЦөФцҙуЈ¬І»Н¬·Ҫ·ЁјЖЛгөДұИЦөУРІ»Н¬ЎЈФЪН¬Т»УҰБҰПВЈ¬FlynnөДДЈРНјЖЛгөДұИЦөЧоРЎЈ¬2ёц·ҪПтЧФА©ЙўјӨ»оДЬІоҫаЧоҙуЈ»ҝјВЗФӯЧУіЪФҘөДFlynnДЈРНјЖЛгөДҪб№ыҙОЦ®Ј»К№УГөЪТ»РФФӯАнNEB№э¶ЙМ¬ЛСЛч·Ҫ·ЁјЖЛгөДұИЦөЧоҙуЈ¬ЧФА©ЙўјӨ»оДЬөДІоТмРФЧоРЎЎЈ

Нј9 іЪФҘәНОҙіЪФҘПВФҫЗЁҫаАлlәНКЖАЭҫаАлyЛжНвБҰөДұд»Ҝ
Fig. 9 Jump distance l and barrier distance y with and without relaxation as function of stress

Нј10 НвБҰУлОЮНвБҰЧчУГПВЧФА©ЙўјӨ»оДЬөДұИЦөЛжУҰБҰөДұд»Ҝ
Fig. 10 Ratio of activation energies with and without stress as function of stress
ҙУ·ЦОцөДҪб№ыҝЙТФҝҙіцЈәFlynnДЈРН№эҙуөДМеПЦБЛУҰБҰ¶ФА©ЙўРРОӘөДУ°ПмЈ¬6%УҰұдПВQ[011]/Q[101]ҙпөҪ0.72Ј¬ХвіэБЛГ»УРҝјВЗФӯЧУіЪФҘәНК№УГКФСйІОКэНвЈ¬Ц»К№УГ¶юО¬ДЈРНјЖЛгТІКЗЦчТӘФӯТтЎЈөЪТ»РФФӯАнөДҪб№ыПВУҰБҰ¶ФА©ЙўРРОӘөДУ°ПмПа¶ФјхРЎЈ¬6%УҰұдПВQ[011]/Q[101]ОӘ0.90Ј¬ІўЗТЛжЧЕУҰБҰөДФцҙуЈ¬УҰБҰ¶ФQ[011]/Q[101]өДУ°ПмЦрҪҘјхРЎЎЈ

Нј11 [011]әН[101]2ёц·ҪПтЙПЧФА©ЙўјӨ»оДЬөДұИЦөЛжЧЕУҰБҰөДұд»Ҝ
Fig. 11 Ratio of activation energies in [011] and [101] directions as function of stress
4 ҪбВЫ
(1) [100]·ҪПтөДөҘЦбАӯЙмУҰБҰФцҙуБЛВБөДҝХО»РОіЙДЬЎЈОЮУҰБҰКұВБөДҝХО»РОіЙДЬОӘ0.695 eVЈ¬ФЪ6%өДУҰұдПВВБөДҝХО»РОіЙДЬФцҙуОӘ0.826 eVЎЈ
(2) ФЪ[100]·ҪПтөДөҘЦбАӯЙмУҰБҰЧчУГПВЈ¬ВБөДЧФА©ЙўјӨ»оДЬФЪ[011]А©Йў·ҪПтЙПјхРЎЈ¬ФЪ[101]А©Йў·ҪПтЙПФцҙуЈ¬УҰБҰөјЦВБЛВБФӯЧУЧФА©ЙўөДёчПтТмРФЎЈОЮУҰБҰЧчУГПВВБөДЧФА©ЙўјӨ»оДЬОӘ1.303 eVЈ¬6%өДУҰұдПВ[011]·ҪПтөДЧФА©ЙўјӨ»оДЬОӘ1.234 eVЈ¬[101]·ҪПтөДЧФА©ЙўјӨ»оДЬОӘ1.371 eVЎЈ
(3) І»Н¬јЖЛг·Ҫ·ЁөГөҪөД[011]А©Йў·ҪПтУл[101]А©Йў·ҪПтөДұИЦөЛжУҰБҰұд»ҜіМ¶ИІ»Н¬ЎЈОҙҝјВЗФӯЧУіЪФҘөДFlynnДЈРНјЖЛгөГөҪөДұИЦөЛжУҰБҰұд»ҜіМ¶ИЧоҙуЈ¬ҝјВЗФӯЧУіЪФҘөДFlynnДЈРНҙОЦ®Ј¬өЪТ»РФФӯАн№э¶ИМ¬ЛСЛч·Ҫ·ЁөГөҪөДҪб№ыЧоРЎЎЈ
ІОҝјОДПЧЈә
[1] Zhu A W, Starke E A. Stress aging of Al-xCu alloys: Experiments[J]. Acta Materialia, 2001, 49: 2285-2295.
[2] іВҙуЗХ, ЦЈЧУйФ, АоКАіҝ, өИ. НвјУУҰБҰ¶ФAl-Cuј°Al-Cu-Mg-AgәПҪрОціцПаЙъіӨөДУ°Пм[J]. ҪрКфС§ұЁ, 2004, 40(8): 799-804.
CHEN Daqin, ZHENG Ziqiao, LI Shichen, et al. Effect of external stress on the growth of precipitate in Al-Cu and Al-Cu-Mg-Ag alloys[J]. Acta Metallurgica Sinica, 2004, 40(8): 799-804.
[3] іВҙуЗХ, АоКАіҝ, ЦЈЧУйФ, өИ. №ІёсіБөнОціц№эіМөДДЈДв. ўт: НвјУУҰБҰіЎөДУ°Пм[J].ЦР№ъУРЙ«ҪрКфС§ұЁ, 2006, 16(1): 116-122.
CHEN Daqin, LI Shichen, ZHENG Ziqiao, et al. Simulations of precipitation process of coherent particles. ўт: Effect of external stress[J]. The Chinese Journal of Nonferrous Metals, 2006, 16(1): 116-122.
[4] НхәкО°, ТЧөӨЗа, ІМҪрБж, өИ. УҰБҰКұР§¶Ф2E12 ВБәПҪрөДБҰС§РФДЬәНОў№ЫЧйЦҜөДУ°Пм[J]. ЦР№ъУРЙ«ҪрКфС§ұЁ, 2011, 21(12): 3019-3025.
WANG Hongwei, YI Danqing, CAI Jinling, et al. Effect of stress aging on mechanical properties and microstructures of 2E12 aluminum alloy[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(12): 3019-3025.
[5] ІЬЛШ·ј, ЕЛЗеБЦ, БхПюСЮ, өИ. НвјУУҰБҰ¶ФAl-Cu-Mg-AgәПҪрКұР§ОціцРРОӘөДУ°Пм[J]. ЦР№ъУРЙ«ҪрКфС§ұЁ, 2010, 20(8): 1513-1519.
CAO Sufang, PAN Qinglin, LIU Xiaoyan, et al. Effects of external stress on aging precipitation behavior of Al-Cu-Mg-Ag alloy[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(8): 1513-1519.
[6] Ardell A J, Prikhodko S V. Coarsening of ҰГЎд in Ni-Al alloys aged under uniaxial compression: II. Diffusion under stress and retardation of coarsening kinetics[J]. Acta Materialia, 2003, 51(17): 5013-5019.
[7] Chen Z Z, Kioussis N, Ghoniem N, et al. Strain-field effects on the formation and migration energies of self interstitials in ҰБ-Fe from first principles[J]. Physical Review B, 2010, 81(9): 094102.
[8] Yashiro K, Oho M, Tomita Y. Ab initio study on the lattice instability of silicon and aluminum under [001] tension[J]. Computational Materials Science, 2004, 29(4): 397-406.
[9] WANG Yunjiang, WANG Chongyu. Influence of the alloying element Re on the ideal tensile and shear strength of ҰГЎд-Ni3Al[J]. Scripta Materialia, 2009, 61(2): 197-200.
[10] Ceperley D M, Alder B J. Ground state of the electron gas by a stochastic method[J]. Physical Review Letters, 1980, 45(7): 566-569.
[11] Kresse G, Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54(16): 11169-11186.
[12] Mills G, Jonsson H. Quantum and thermal effects in H2 dissociative adsorption: Evaluation of free energy barriers in multidimensional quantum systems[J]. Physical Review Letters, 1994, 72(7): 1124-1127.
[13] Wang C, Han P D, Zhang L, et al. The strengthening effect of Al atoms into Mg-Al alloy: A first-principles study[J]. Journal of Alloys and Compounds, 2009, 482(1/2): 540-543.
[14] Lu G H, Deng S H, Wang T M. Theoretical tensile strength of an Al grain boundary[J]. Physical Review B, 2004, 69(13): 134106.
[15] КҰІэРч. ІДБПҙуҙКөд[M] . ұұҫ©:»ҜС§№ӨТөіц°жЙз, 1994: 1060-1061.
SHI Changxu. Materials dictionary[M]. Beijing: Chemical Industry Press, 1994: 1060-1061.
[16] Carling K, Wahnstrom G. Vacancies in metals: from first-principles calculations to experimental data[J]. Physical Review Letters, 2000, 85(18): 3862-3865.
[17] Sandberg N, Magyari-kope B, Mattsson T R. Self-diffusion rates in Al from combined first-principles and model-potential calculations[J]. Physical Review Letters, 2002, 89(6): 065901.
[18] Mantina M, Wang Y, Arroyave R, et al. First-principles calculation of self-diffusion coefficients[J]. Physical Review Letters, 2008, 100(21): 215901.
[19] Flynn C P. Atomic migration in monatomic crystals[J]. Physical Review, 1968, 171(3): 682-698.
(ұајӯ СоУЧЖҪ)
КХёеИХЖЪЈә2012-06-04Ј»РЮ»ШИХЖЪЈә2012-10-21
»щҪрПоДҝЈә№ъјТЧФИ»ҝЖС§»щҪрЧКЦъПоДҝ(51071177)
НЁРЕЧчХЯЈәТЧөӨЗа(1954-)Ј¬ДРЈ¬әюДППжПзИЛЈ¬ҪМКЪЈ¬І©КҝЙъөјКҰЈ¬ҙУКВёЯРФДЬЗбәПҪрј°·ЫД©ТұҪрРВІДБПСРҫҝЈ»өз»°Јә0731-88830263Ј»E-mailЈәyioffice@ csu.edu.cn

