文章编号:1004-0609(2009)08-1505-06
Au/AC催化剂的制备及其催化性能
张 博1,张彭义1,徐九华2,李宏煦3,史 蕊3
(1. 清华大学 环境科学与工程系,北京 100084;
2. 北京科技大学 土木与环境工程学院,北京 100083;
3. 北京科技大学 冶金与生态工程学院,北京 100083)
摘 要:采用浸渍法制备改性活性炭负载金催化剂(Au/AC),并通过N2吸附?脱附、光电子能谱(XPS)对该催化剂进行表征,考察Au/AC催化剂在室温下对低浓度臭氧、甲苯以及二者共存时的催化分解性能。结果表明:Au/AC催化剂对臭氧具有优良的催化分解活性,在室温、空速76 000 h?1、相对湿度(45±5)%条件下,Au/AC对初始浓度为(55±5) mg/m3 的臭氧在2 300 min时的去除率为91.3%;Au/AC对甲苯的去除未表现出明显的催化分解性能,而是以吸附作用为主;在臭氧和甲苯共存的条件下,Au/AC对臭氧的催化分解活性显著降低;与臭氧反应后,Au/AC的表面石墨碳含量显著降低,说明臭氧能够通过Au/AC催化其与活性炭的反应得以降解,且在反应前后,活性组分Au能够保持稳定的单质价态。
关键词:金催化剂;催化;臭氧;浸渍法
中图分类号:O 643 文献标识码: A
Preparation and catalytic activity of Au/AC catalyst
ZHANG Bo1, ZHANG Peng-yi1, XU Jiu-hua2, LI Hong-xu3, SHI Rui3
(1. Department of Environmental Science and Engineering, Tsinghua University, Beijing 100084,China;
2. School of Civil and Environmental Engineering, University of Science and Technology Beijing,
Beijing 100083, China;
3. School of Metallurgical and Ecological Engineering, University of Science and Technology Beijing,
Beijing 100083,China)
Abstract: The catalysts of Au supported on modified activated carbon were prepared by impregnation method and characterized by N2 adsorption-desorption and XPS. The catalytic activity of Au/AC for decomposition of ozone and toluene was evaluated. The results show that, under the condition of (55±5) mg/m3 ozone in air, space velocity 76 000 h?1, relative humidity (45±5)% and ambient temperature, the ozone removal ratio of the catalyst can be maintained at 91.3% within 2 300 min. And the activity of Au/AC catalyst is reduced when the toluene exists in the air. The graphitic carbon content of Au/AC decreases dramatically, which indicates that Au/AC can promote the reaction betweens the ozone and carbon. For the reason of competitive adsorption when ozone and toluene coexist, the activity of Au/AC declines obviously. However, Au maintains its stable state of metallic gold before and after the reaction.
Key words: gold catalyst; catalysis; ozone; impregnation
自20世纪80年代后期,HARUTA等[1]发现负载在过渡金属氧化物上的纳米金催化剂对低温CO氧化具有较高的催化活性后,人们掀起了对纳米金催化剂的研究热潮。近年来,载金催化剂在低温CO氧化[2?5],烯、醇类有机物环氧化[6?7]、甲烷催化燃烧[8?9]以及低温水气反应[10]等方面的催化活性受到广泛关注。HAO等[11]和林清泉等[12]分别研究了金属氧化物载金催化剂对臭氧和CO的去除效果。ROSSI等[13]和CARRETTIN等[13?14]分别制备了负载量为0.8%的X40S型椰壳活性炭载金催化剂和负载量为1%的XC72R型石油脑活性炭载金催化剂,并考察了它们对糖和乙醇液相氧化的催化活性。但活性炭载金催化剂在室温下对低浓度臭氧的催化分解活性、以及甲苯对其活性的影响还鲜见报道。本文作者以降低居室内的臭氧浓度为目的,并考虑室内空气的复杂性而加入挥发性有机污染物甲苯,研究以改性活性炭为载体,采用简单易行的浸渍法制备Au/AC催化剂,考察这种催化剂对臭氧的催化分解性能、以及甲苯的介入对其催化活性的影响。
1 实验
1.1 催化剂的制备
以山西新华化工厂生产的DX09型煤质活性炭(记为AC)为载体,对其进行改性处理:活性炭经过10%的稀硝酸在60 ℃水浴中煮2 h,再用2.5 mol/L NaBH4室温下浸泡4 h后,洗至中性并烘干备用(记为M-AC)。以氯金酸(HAuCl4・4H2O)为金前驱体溶液,乙二胺(En)为配合物,将其按质量比1?1配制金含量为 5 g/L的水溶液作为浸渍液。将5 g改性后的活性炭浸渍到10 mL浸渍液中,超声处理2 min使其分散均匀,过滤出活性炭在常温下晾干,并在200 ℃下用H2还原40 min,即制得活性炭载金催化剂(记为Au/AC)。
1.2 催化剂的表征
采用氮吸附?脱附仪(QuadraSorb SI,氮气为吸附质,相对压力p/p0为10?6~1)测定催化剂的比表面积和孔隙结构。采用光电子能谱仪(PHI-5300 ESCA,Al/Mg为双阳极靶,采用C1s(能量为284.8 eV))进行荷电校正,测定催化剂表面的元素组成及价态。
1.3 催化剂的性能测试
催化剂活性测试在常温常压下,采用填充床连续流动方式进行评价。在内径为2 cm的不锈钢管内装入1.6 g催化剂,床层厚度为0.8 cm,空气流速为4 L/min,对应的空速为96 000 h?1,接触时间为0.04 s。臭氧进口浓度为(55±5)mg/m3,气流的相对湿度为(45±5)%。臭氧浓度及相对湿度分别由美国热电公司的Model 49C型臭氧分析仪、Mannix Lan 880D型温湿度计测定。催化剂的活性以其对臭氧的去除率以及持续时间来表示。甲苯初始浓度为3.8 mg/m3,由气相色谱仪HP5890H/PH分析测定,通过六通阀直接进样,进样量为0.5 mL,色谱条件如下:填充柱为OV?101,载气N2的压力为80 kPa,进样口温度150 ℃,柱温90 ℃,检测器温度200 ℃。
2 结果与讨论
2.1 催化剂活性评价
2.1.1 Au/AC对臭氧催化分解活性
图1所示为AC、M-AC及Au/AC这3种样品对臭氧的分解性能。比较三者对臭氧的去除率曲线可知,Au/AC催化剂对臭氧的催化分解性能明显优于载体AC和M-AC的催化分解性能,在1 400 min时,Au/AC的去除率保持在95%以上,而此时AC和M-AC对臭氧的去除率已分别下降到72%和78%。由此可见,负载活性组分金制成的Au/AC催化剂与未负载的AC和M-AC相比,Au/AC对臭氧的去除性能大幅度提高,说明负载的活性组分金具有催化分解臭氧的活性。对比AC及M-AC对臭氧的去除曲线可知,M-AC对臭氧的分解性能略优于AC的,说明对AC进行改性处理有利于提高对臭氧的去除能力。

图1 AC、M-AC及Au/AC对臭氧的分解性能
Fig.1 Decomposition ability of AC, M-AC and Au/AC catalyst for ozone
2.1.2 Au/AC对甲苯的去除性能
实验中考察了在无臭氧存在时Au/AC对甲苯的去除能力。图2所示为AC、M-AC和Au/AC对甲苯的去除能力。由图2可以看出,3条曲线的下降趋势类似,但明显不同于图1中3条臭氧去除曲线所表现出的随时间推移缓慢下降的趋势。在本组试验中,反应前期3种样品对甲苯均保持较高的去除效率,且呈现平缓下降的趋势;但随着时间的延长,在某一时刻去除率突然急速下降至20%左右。这种去除曲线所表现出的特征更符合固?气相吸附穿透曲线的特性,因此,在此反应中,吸附作用应大于催化分解作用。AC在前1 051 min内对甲苯的去除能力属平缓下降期,此时AC对甲苯的去除性能优于M-AC和Au/AC的,其去除效率由100%平缓下降至96%;但1 051 min后,甲苯去除率迅速下降,在2 054 min时已下降到15%。而M-AC及Au/AC去除甲苯的平缓下降期均有所延长,分别在1 364 min和1 890 min后急速下降。AC、M-AC和Au/AC在近1 850 min时对甲苯的去除率分别下降至38.4%,25.1%和83.3%,估算在此时间内3种样品对甲苯的去除量分别为17.16,17.31和26.32 mg。这说明Au/AC对甲苯的去除虽然以吸附作用为主,但其去除甲苯能力强于负载前的AC和M-AC,负载的金颗粒具有一定催化分解甲苯的能力。
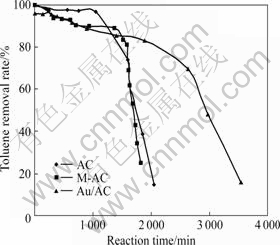
图2 AC、M-AC及Au/AC对甲苯的去除性能
Fig.2 Decomposition ability of AC, M-AC and Au/AC catalysts for toluene
2.1.3 臭氧与甲苯共存时Au/AC的催化活性测试
图3所示分别为臭氧和甲苯共存时AC、M-AC及Au/AC对两种污染物的去除率曲线。由图3(a)可知,在有甲苯介入时,3种样品对臭氧的去除率曲线表现出一致的下降趋势,与图1所示的结果相比,3种样品对臭氧的去除能力都显著下降,尤以Au/AC为甚, Au/AC并未表现出对臭氧的催化分解性能远优于AC和M-AC的趋势,说明甲苯的介入不利于Au/AC对臭氧的催化分解,降低了Au/AC的催化活性。对比图3(b)中的3条甲苯去除率曲线可以看到,三者的下降趋势明显不同。AC和M-AC甲苯去除率曲线的下降趋势与图2的3条曲线类似,仍表现出固?气相吸附穿透曲线的特性,说明在臭氧和甲苯共存的条件下,AC与M-AC对甲苯的去除仍以吸附作用为主。但Au/AC的甲苯去除率曲线下降趋势却发生变化,随反应时间的增加甲苯的去除率一直保持平缓下降趋势,这说明在两种污染物质共存条件下,Au/AC对甲苯的去除已由单一甲苯存在时以吸附作用为主,转变为以催化分解作用为主。由以上测试结果可以认为,当有甲苯介入时,由于甲苯和臭氧对Au/AC的金活性位存在竞争吸附作用,导致吸附并催化分解臭氧的有效活性位数量大量减少,使其对臭氧的分解性能显著下降。同时Au/AC的元素组成和孔隙结构由于臭氧的催化分解反应而发生变化,使其对甲苯的吸附性能也大大降低。在臭氧和甲苯共存条件下,Au/AC能够促进一部分甲苯与臭氧发生反应,导致甲苯的主要去除机制发生改变。

图3 臭氧与甲苯共存条件下AC、M-AC及Au/AC对二者的去除性能
Fig.3 Decomposition ability of AC, M-AC and Au/AC catatysts for ozone (a) and toluence (b) at coexisting ozone and toluene
2.2 催化剂表征
2.2.1 N2吸附?脱附表征
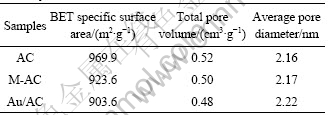
对AC、M-AC和Au/AC这3种样品进行了N2吸附?脱附表征,根据N2吸附?脱附等温线计算得到各样品的比表面积、孔容和孔径,结果列于表1。由表1可看出,样品AC、M-AC和Au/AC的BET比表面积和总孔容均依次下降,而平均孔径逐渐升高。这是由于AC在改性过程中,其孔隙结构遭到破坏,造成部分孔隙发生结构坍塌现象,使比表面积和总孔容下降。负载金以后的Au/AC比表面积和总孔容进一步下降,考虑到金的原子半径为0.144 nm[15],而孔径为2 nm以下微孔在活性炭载体的孔隙组成中占有较大比例。因此,可以推断,部分纳米金颗粒进入到载体的微孔中导致部分微孔堵塞,而微孔对比表面积和总孔容的贡献最大,所以当微孔数量减少时,必然导致比表面积和总孔容下降而平均孔径增加。
表1 载体与催化剂样品的N2吸附?脱附表结果
Table 1 Results from N2 adsorption-desorption measurement of catalysts AC, M-AC and Au/AC
2.2.2 XPS表征
分别对未参加反应的催化剂Au/AC、与臭氧反应后的Au/AC(简称Au/AC/O3)、与甲苯反应后的Au/AC(简称Au/AC/C7H8)以及在臭氧、甲苯共存条件下反应后的Au/AC(简称Au/AC/(O3+C7H8)) 4种样品进行XPS表征,通过对样品表面含氧官能团相对百分含量变化情况的考察,进一步分析Au/AC的催化作用机制。C1s结合能谱峰可解析为5种独立的峰,分别为:石墨碳,酚、醇和醚的C―OH,羰基和醌的C=O,羧基和酯的COOR以及π-π*电子携上线[16?17]。各样品的解析结果如表2所列。比较4种样品中含氧官能团的相对含量可以看出,与未参加反应的Au/AC相比,在其它3种反应后的样品中,Au/AC/C7H8各官能团的相对含量变化幅度最小,催化剂表面几乎没有发生反应,结合图2中对甲苯去除率曲线特点的分析,进一步证明了Au/AC对甲苯的去除是以吸附作用为主。Au/AC/O3各官能团相对含量变化最大,其中石墨的相对含量碳由70.1%下降到54.1%,COOR的相对含量碳由4.3%上升到13.9%,说明在臭氧催化分解反应过程中随反应时间增加,石墨碳、C―OR、C=O被不断氧化,氧化产物COOR由于不易被进一步氧化成CO或CO2而有所累积,导致其含量大幅度增加。Au/AC/(O3+C7H8)样品中各官能团的相对含量变化幅度介于前两者之间,原因是当有甲苯介入时,臭氧和甲苯对Au/AC表面的活性位存在竞争吸附,一部分活性位由于吸附了甲苯而丧失对臭氧的催化分解活性,使催化臭氧分解反应的有效活性位数量减少,从而导致与臭氧发生反应的碳元素的量也相应减少。因此,在两种污染物共存的条件下,与Au/AC/O3相比,Au/AC/(O3+C7H8)的表面石墨碳的消耗量减少,对臭氧的去除性能下降。同时,催化剂在催化分解臭氧反应过程中自身表面元素组成和孔隙结构的改变,导致Au/AC/(O3+C7H8) 对甲苯的吸附性能也远低于Au/AC/C7H8的。
图4所示为4种样品表面Au4f的XPS能谱。Au/AC样品表面Au4f7/2的结合能位置为84.2 eV,对应的元素化学态为单质金[18]。由图4可看出,经过3种不同反应过程后,样品Au/AC/O3、Au/AC/C7H8 和Au/AC/(O3+C7H8)表面Au 4f的结合能并没有发生明显位移,说明活性组分Au在反应前后能够保持稳定的单质价态。因此,结合性能测试结果和表征结果可以认为,Au/AC能够通过催化臭氧与活性炭的反应来实现对臭氧的降解,且在反应过程中Au能够保持稳定的单质价态。
表2 不同样品的C1s XPS谱拟合结果
Table 2 Fitting results of C1s XPS spectra of different samples


图4 不同样品的Au4f XPS谱
Fig.4 Au4f XPS spectra of different samples
3 结论
1) 以乙二胺为配体保护剂采用浸渍法制备的改性活性炭载金催化剂,在室温条件下,催化分解浓度为(55±5)mg/m3的臭氧,在2 300 min时,对臭氧的去除率为91.3%。XPS数据显示,Au/AC在催化臭氧分解反应后,其表面的石墨碳相对百分含量明显降低,活性组分金在反应前后始终保持稳定的单质状态,说明Au/AC能够通过催化臭氧与活性炭的反应来实现对臭氧的降解。
2) 在臭氧与甲苯共存的条件下,由于甲苯的介入使Au/AC用于催化分解臭氧的有效活性位数量减少,导致Au/AC对臭氧的去除率明显降低。同时,臭氧与碳的反应又破坏了Au/AC的表面元素组成和孔隙结构从而降低了对甲苯的吸附性能。
REFERENCES
[1] HARUTA M, YAMADA N, KOBYSAHI T, LIJIMA S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide[J]. J Catal, 1989, 115(2): 301?309.
[2] BULUSHEV D A, YURANOV I, SUVOROVA E I, BUFFAT P A, KIWI-MINSKER L. Highly dispersed gold on activated carbon fibers for low-temperature CO oxidation[J]. J Catal, 2004, 224(1): 8?17.
[3] 齐世学, 邹旭华, 刘雪梅, 安立敦. Au/TiO2催化剂的制备及其催化性能[J]. 化学通报, 2005(10): 785?788.
QI Shi-xue, ZOU Xu-hua, LIU Xue-mei, AN Li-dun. The preparation and photocatalytic activity of Au/TiO2[J]. Chemistry, 2005(10): 785?788.
[4] PILLAI U R, DEEVI S. Highly active gold-ceria catalyst for the room temperature oxidation of carbon monoxide[J]. Appl Catal A, 2006, 299(2): 266?273.
[5] DAT? M, IMAI H, TSUBOTA S, HARUTA M. In situ measurements under flow condition of the CO oxidation over supported gold nanoparticles[J]. Catal Today, 2007, 122(3/4): 222?225.
[6] CHOWDHURY B, BRAVO-SUAREZ J J, DATE M, TSUBOTA S, HARUTA M. Trimethylamine as a gas-phase promoter: Highly efficient epoxidation of propylene over supported gold catalysts[J]. Angew Chem, 2005, 118(3): 426?429.
[7] DIMITRATOS N, LOPEZ-SANCHEZ J A, LENNON D, PORTA F, PRATI L, VILLA A. Effect of particle size on monometallic and bimetallic (Au, Pd)/C on the liquid phase oxidation of glycerol[J]. Catal Lett, 2006, 108(3/4): 147?153.
[8] WATERS R D, WEIMER J J, SMITH J E. An investigation of the activity of coprecipitated gold catalysts for methane oxidation[J]. Catal Lett, 1995, 30(1/4): 181?188
[9] MIAO S J, DENG Y Q. Au-Pt/Co3O4 catalyst for methane combustion[J]. Appl Catal B, 2001, 31(3): L1?L4.
[10] 华金铭, 郑 起, 林性贻, 魏可镁. 金负载量对低温水煤气变换Au/α-Fe2O3催化剂结构和性能的影响[J]. 燃料化学学报, 2003, 31(6): 558?563.
HUA Jin-ming, ZHENG Qi, LIN Xing-yi, WEI Ke-mei. Influence of Au loading on the structure and catalytic performance of Au/α-Fe2O3 catalysts for low temperature water-gas shift reaction[J]. Journal of Fuel Chemistry and Technology, 2003, 31(6): 558?563.
[11] HAO Z P, CHENG D Y, GUO Y, LIANG Y H. Supported gold catalysts used for ozone decomposition and simultaneous elimination of ozone and carbon monoxide at ambient temperature[J]. Appl Catal B, 2001, 33(3): 217?222.
[12] 林清泉, 安立敦, 陈俊勇, 秦 华, 齐世学, 邹旭华. LaFeO3修饰和O3处理对Au/Al2O3催化剂在CO氧化反应中热稳定性的影响[J]. 催化学报, 2008, 29(6): 506?508.
LIN Qing-quan, AN Li-dun, CHEN Jun-yong, QIN Hua, QI Shi-xue, ZOU Xu-hua. Effect of LaFeO3 decoration and ozone treatment on the thermal stability of Au/Al2O3 for CO oxidation [J]. Journal of Catalyst, 2008, 29(6): 506?508.
[13] PORTA F, PRATI L, ROSS MI, SCARI G. New Au(0) sols as precursors for heterogeneous liquid-phase oxidation catalysts[J]. J Catal, 2002, 211(2): 464?469.
[14] CARRETTIN S, MCMORN P, JOHNSTON P, GRIFFIN K, HUTCHINGS G J. Selective oxidation of glycerol to glyceric acid using a gold catalyst in aqueous sodium hydroxide[J]. Chem Commun, 2002, 2(7): 696?697.
[15] DEAN J A. Lange’s handbook of chemistry[M]. 15th Ed. New York: McGraw-Hill Press, 1999: 31.
[16] WU S H, PENDLETON P. Adsorption of anionic surfactant activated carbon: Effect of surface chemistry, ionic strength, and hydrophobicity[J]. J Colloid Interface Sci, 2001, 243(2): 306?315.
[17] SUBRAHMANYAM C, BULUSHEV DA, KIWI-MINSKER L. Dynamic behaviour of activated carbon catalysts during ozone decomposition at room temperature[J]. Appl Catal B, 2005, 61(1/2): 98?106.
[18] MINIC? S, SCIR? S, CRISAFULLI C, GALVAGNO S. Influence of catalyst pretreatments on volatile organic compounds oxidation over gold/iron oxide[J]. Appl Catal B, 2001, 34(4): 277?285.
基金项目:国家自然科学基金资助项目(50772058);国家重点实验室专项基金资助项目(08Y02ESPCT)
收稿日期:2008-11-10;修订日期:2009-04-17
通讯作者:张彭义,教授,博士;电话:010-62796840-601;E-mail: zpy@tsinghua.edu.cn
(编辑 龙怀中)

