苯并噻(噁)唑酮衍生物杀虫活性的理论研究
冯长君1,刘志国2,梁逸曾3
(1. 徐州工程学院 化学化工学院,江苏 徐州,221111;
2. 湖南工业大学 绿色包装与生物纳米技术应用湖南省重点实验室,湖南 株洲,412008;
3. 中南大学 化学化工学院,湖南 长沙,410083)
摘要:用DFT-B3LYP方法,在基组6-31G水平下, 对34种N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺类化合物分子进行几何及电子结构全优化,将ELUMO(最低空轨道能),EHOMO(最高占据轨道能),ENLUMO(次最低轨道能),ENHOMO(次最高占据轨道能),△E1(=ELUMO-EHOMO),△E2(=ENLUMO-ENHOMO), QZ(分子中原子的净电荷),μ(偶极矩),Ev(零点振动能),E(热力学能),Cv(比热容),Sm(熵)和前沿分子轨道组成及其电子密度等参数被选作量子化学描述符(Lc)。通过最佳变量子集回归建立15种上述化合物对蚕豆蚜和棉红蜘蛛等杀虫活性(SJ:Sa,Su)的QSAR模型。蚕豆蚜的Sa模型的相关系数(R2)和逐一剔除法交叉验证系数Rcv2依次为0.897和0.858, 棉红蜘蛛Su模型的相应参数为0.858和0.757。经Radj2,F,Rcv2,VIF和FIT等检验, 上述模型具有较强的稳健性和预测能力。根据模型可知,这些化合物杀虫机理包含2点:一是与生物体内大分子之间的电子转移反应,以△E1和ENLUMO来描述;二是氢键,用QN2表述。其中,ENLUMO与杀虫活性正相关,而△E1和QN2升高,SJ下降。设计与预测19种新颖化合物的杀虫活性,其中2种化合物的杀虫活性均超过100%(质量浓度为500 mg/dm3)。所建模型可为实验工作者合成新型高效N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺类化合物提供理论参考。
关键词:N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺类化合物;密度泛函理论(DFT);量化参数;蚕豆蚜;棉红蜘蛛;杀虫活性;杀虫机理;定量构效关系(QSAR)
中图分类号:S482.3+7;O6-051 文献标志码:A 文章编号:1672-7207(2013)11-4399-09
Theoretical study on insecticidal activities and structural modification for N-arylaminocarbonyl-2-benzothiazolinone/benzoxazolone
FENG Changjun1, LIU Zhiguo2, LIANG Yizeng3
(1. School of Chemistry & Chemical Engineering, Xuzhou Institute of Technology, Xuzhou 221111, China;
2. Key Laboratory of Green Packaging and Application Biological Nanotechnology of Hunan Province, Hunan University of Technology, Zhuzhou 412008, China;
3. College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China)
Abstract: The density functional theory (DFT)/B3LYP method, with the basic set 6-31G, was employed to calculate the molecular geometries and electronic structures of 34 N-arylaminocarbonyl-2-benzothiazolinone/benzoxazolone compounds. ELUMO, EHOMO, ENLUMO, ENHOMO, △E1(=ELUMO-EHOMO), △E2(=ENLUMO-ENHOMO), QZ (the net charge of atoms in molecules), μ (dipole moment), Ev (vibrational energy of 0 K), E(thermodynamic energy), Cv (heat capacity), Sm (entropy), constituents of frontier molecular orbitals and their atomic frontier electron densities were selected as structural descriptors (Lc). The insecticidal activities (SJ:Sa, Su) of 15 compounds among the above-mentioned compounds to Aphis fabae, Tetranychus urticae along with the above structural parameters, was used to established the quantitative structure-activity relationships (QSAR) by using leaps-and-bounds regression analysis. The correlation coefficients (R2) and the leave-one-out (LOO) cross validation Rcv2 for the Sa and Su models were 0.897 and 0.858, 0.858 and 0.757, respectively. The QSAR models have both favorable estimation stability and good prediction capability by Radj2, F, Rcv2, VIF, FIT tests. In light of the model, the insecticidal mechanism of these compounds may include two points, one is the electron transfer reactions between the chemicals and macromolecular compounds in vivo described by △E1 and ENLUMO; and the other is the hydrogen bonds represented by QN2. The insecticidal activities of the compounds increase with the increase of the ENLUMO. The higher the △E1 and QN2 is, the lower the SJ is. The insecticidal activity parameters of 19 new compounds were designed and predicted using the QSAR models, of which two were disclosed to have better insecticidal activities (the insecticidal activities are over 100% when concentration is 500 mg/dm3). The models may be used as a theoretical reference for the design of new N-arylaminocarbonyl-2-benzothiazolinone/benzox-azolone compounds.
Key words: N-arylaminocarbonyl-2-benzothiazolinone/benzoxazolone; density functional theory (DFT); quantum chemical descriptor; Aphis fabae; Tetranychus urticae; insecticidal activity; insecticidal mechanism; quantitative structure-activity relationship (QSAR)
具有极高生物活性的稠杂环化合物已受到人们极大关注,其中苯并噻唑与苯并噁唑衍生物具有广泛的生物活性(表现为杀虫、杀菌、抗病毒、除草、植物生长调节等活性)[1],在新农药创制中起到重要作用。翁建全等[2]根据生物活性叠加原理,利用苯并噻(噁)唑酮与取代苯基异氰酸酯的偶合反应, 合成15个N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺类化合物(N- Arylaminocarbonyl-2-benzothiazolinone/benzoxazolone compounds,简称苯并噻(噁)唑酮衍生物)。杀虫测试表明:这些化合物对蚕豆蚜和棉红蜘蛛等具有较强的杀虫活性[2]。物质构效关系(QSAR)[3-7]研究主要是利用各种统计分析工具建立化合物的结构与其性能(如理化性质、生物活性等)之间的数学关系,以此预测其他化合物的相关性质;并在分子水平上揭示微观结构对化合物各种生物活性的影响,推测其可能的作用机理。为进一步提高上述N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺化合物的杀虫活性,本文作者通过包含电子相关的密度泛函理论(DFT) B3LYP方法[8-10]对其进行构效关系研究。即从几何构型、电荷分布和前线轨道能等方面探讨该类化合物的结构与杀虫活性之间的关系,采用最佳变量子集回归(LBR)程序建立它们与蚕豆蚜、棉红蜘蛛等杀虫活性(SJ)[2]的数学模型,并获得影响该类化合物杀虫活性的主要因素,进而推测其与受体作用的杀虫机理,据此设计出2个对蚕豆蚜、棉红蜘蛛具有很高杀虫活性的新的N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺类化合物, 期望能为将来相关的生物实验所证实,而开发出高效杀虫剂。
1 理论与方法
1.1 杀虫活性数据
15种N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺化合物的基本结构是(图1),相应取代基(R,即分子中可变部分)如表1所示。表1中19个加“*”号的为新设计的化合物。采用浸渍法对标题化合物进行蚕豆蚜和棉红蜘蛛杀虫活性的测定,质量浓度为500 mg/L,并设空白对照(CK)。这些化合物杀虫活性以“死亡率(SJ)”表示[2],具体数据如表1所示,Sa和Su依次代表致使蚕豆蚜、棉红蜘蛛的死亡率。

图1 标题化合物的基本结构
Fig.1 Basic structure of title compounds
1.2 量子化学参数计算
由于该类化合物分子均无任何对称性,为节省计算时间,先用Chem3D程序构建分子,再用其中MoPAC的AM1程序对各化合物分子结构进行初步几何优化。然后,运用G98量子化学程序包,采用密度泛函理论(DFT)的B3LYP(即Becke三参数混合泛函)方法,在6-31G 基组水平上,按闭壳层单重态进行全几何优化及频率计算。同时经振动分析,所得稳定构型均无虚频,表明对应于各自势能面上的极小点,不是马鞍点,所得量化参数均对应分子的最稳定构型。根据前线分子轨道理论,电子的跃迁是由反应物分子前线轨道之间的相互作用引起的,前线轨道对化学反应起决定的作用。当然,也对生物体内的生物化学反应发挥主导作用。因此,选取最高占据轨道能(EHOMO)、次最高占据轨道能(ENHOMO)、最低空轨道能(ELUMO)、次最低空轨道能(ENLUMO)以及二者轨道能隙△E1(=ELUMO-EHOMO)和△E2(=ENLUMO-ENHOMO)作为表征分子间相互作用的参数。由于EHOMO越高,越易给出电子,还原性越强;ELUMO越低,越易接受电子,氧化性越强,因此,△E1越小,其化学活泼性越强。根据传统化学理论,所有化学反应都是电荷或轨道间相互作用引起的,分子内的电荷是引起电子相互作用的主要原因。因此,计算所有分子中都共有的原子的电荷,即图1中除Y和R外的全部非氢原子的电荷,依次表示为:QC1-QC6(左边苯环中碳原子电荷),QC9-QC15(右边苯环中碳原子电荷),QO(醚氧原子上的电荷),QO1(环中羰基氧原子上的电荷),QO2(非环羰基氧原子上的电荷),QN1-QN2,QC7-QC8(2个苯环之间的碳原子电荷)和QS。上述原子的电荷均以“QZ”表示。偶极矩(μ)是衡量分子整体极性强弱的重要参数,也作为量子化学描述符(Lc)之一。考虑构建QSAR模型的需要,还选取分子的零点振动能(Ev)、热力学能(E)、热容(Cv)及熵(Sm)等。上述所有参数均以“Lc”表示。所有计算都利用Gaussian 98程序微机上完成,计算的收敛精度均采用程序内定值。
表1 N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺化合物的杀虫活性(SJ)与量化参数(Lc)
Table 1 Insecticidal activities (SJ) and quantum chemical parameters (Lc) of N-Arylaminocarbonyl-2-benzothiazolinone/benzoxazolone compounds

1.3 统计回归分析
将每种化合物的上述量化参数(Lc)作为自变量集,相应的2种杀虫活性(Si)分别为因变量,应用最佳变量子集回归选择最佳变量组合,建立相应定量结构-抑菌活性的QSAR模型。采用逐一剔除法(LOO)对模型的预测能力及稳健性进行检验,以交叉验证相关系数(Rcv2)予以评价。一般认为Rcv2≥0.5,所建模型具有良好的稳健性与预测能力。用变异膨胀因子(VIF)[7]评价模型中各自变量的多重相关性,VIF的计算式为
VIF=1/(1-β2) (1)
其中:β2为自变量X中某一变量与余下变量的判定系数。如VIF=1,表明各自变量间完全不相关; 当VIF<5时,说明变量间没有明显的自相关性,所建模型是稳定的;当VIF>5时,说明变量间存在明显的共线性,所建模型不能用于估算与预测。
为确立最终的QSRR模型,引入Akaike信息判据(AIC),Kubinyi函数(FIT)[11-12],其计算公式分别如下:
 (2)
(2)
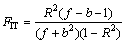 (3)
(3)
其中:RSS为方差和;f为模型所含的化合物数;b为模型中的变量数;R2为因变量与自变量之间的判定系数,即相关系数的平方。AIC越小,FIT越大,所建的模型越稳定,预测能力越强。
2 SJ的QSAR分析
2.1 SJ的多元线性回归模型的确立
QSAR 是从化合物的结构出发建造数学模型,然后运用这种模型去预测化合物的活性或性质,从而为新分子的设计、评价提供理论依据。将15种N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺化合物的自变量(Lc)及因变量(SJ)所构建的数据集输入MINITAB统计分析软件,运用其中的最佳子集回归选择最佳变量组合,建立相应的QSAR模型。标题化合物对蚕豆蚜死亡率Sa的最佳变量组合如表2所示,其中R2,Radj2,Rcv2,S,F,AIC和FIT分别为判定系数(亦称削减误差比例)、校正判定系数(以消除自变量个数及样本容量对判定系数的影响)、逐一剔除法的交叉验证系数、估计标准误差、Fisher统计值、Akaike信息判据、Kubinyi函数。
表2 Lc与Sa的最佳子集回归结果
Table 2 Results of quantum chemical descriptors(Lc) and Sa with leaps-and-bounds regression
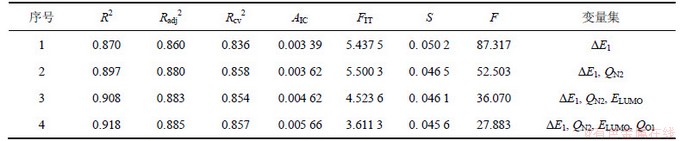
在多元回归分析中,为使所得预测模型具有较高的可信度,一般遵循如下经验规则:f/b≥5[13]。本研究的样本容量f=15,其中b≤3,因此,可建立三元数学模型。由表2可知,随着自变量数目增多,其R2,Radj2和AIC逐渐增大,而F和S逐渐减小,显然不能按此5种指标来确定数学模型。由于Rcv2与FIT 均在二元数学模型处出现转折,即此模型质量相对最好,为蚕豆蚜杀虫活性的最佳QSAR模型:
Sa=1.948-14.596△E1-1.240QN2
f=15, R2=0.897, Radj2=0.880, P=0, Rcv2=0.858,
S=0.046 5, F=52.503 (4)
同理,建立对棉红蜘蛛杀虫活性的最佳QSAR模型:
Su=3.600-17.120△E1+6.357ENLUMO
f =15, R2=0.858, Radj2=0.835, P=0, Rcv2=0.757,
S=0.065 9, F=36.361 (5)
上述杀虫活性的实验值与模型预测值(表1)基本吻合。
2.2 SJ的多元线性回归模型的检验
Yao等[14]提出,若要建立一个具有良好预测能力的QSAR模型,其R2≥0.80。模型(式(4))的R2=0.897,模型(式(5))的R2=0.858,分别反映影响其杀虫活性的89.7%和85.8%因素,仅有10.3%及14.2%的随机因素未被揭示,因此,具有良好预测能力。模型(式(4))的QN2和△E1的VIF为2.275,模型(式(5))的ENLUMO和△E1的VIF为1.373,都小于5且很接近1,说明这些模型中基本不存在共线性,是高度稳定的。又根据Rcv2(≥0.5),F和FIT可知,它们都具有良好的稳健性与预测能力,不仅可用于标题化合物杀虫活性的估算与预测,也可用于杀虫机理的探讨,并进行结构修饰,以获得高活性新颖的化合物。
为进一步验证所建QSAR模型的预测能力,把15个化合物分成训练集12个化合物和测试集3个化合物,共形成455个训练集与测试集。利用MATLAB自编程序计算这些训练集、测试集的相关系数及逐三剔除法(LTO)的交叉验证相关系数(Rcv-32)。对于455个Sa训练集二元方程的相关系数的平均值为0.949,相应Rcv-32 = 0.852;455个Su训练集二元方程的相关系数的平均值为0.926,相应Rcv-32 = 0.751。它们的相关系数平均值及Rcv-32与模型(4)~(5)非常接近。如其中剔除序号为1, 7, 12这3个化合物为测试集,余下为训练集,所建立的模型如下:
Sa=1.587-13.460△E1-1.436QN2
N=12, R2=0.887, R=0.942, Radj2=0.862, P=0,
S=0.045 7, F=35.469 (6)
Su=3.927-18.870△E1+6.115ENLUMO
N=13, R2=0.850, R=0.922, Radj2=0.817, P=0,
S=0.068 6, F=5.561 (7)
模型(6)和(7)给出的计算值及测试集的预测值分别如图2和3所示,可见它们都具有较好的线性相关性。

图2 标题化合物对蚕豆蚜杀虫活性的实验值与计算值的关系
Fig.2 Relationship between experimental and calculated insecticidal activities of title compounds to Aphis fabae
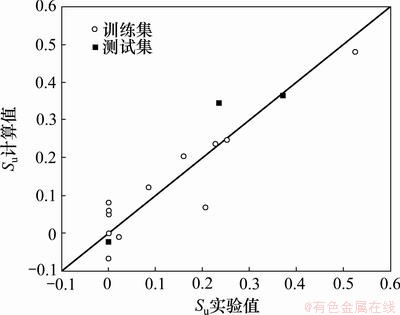
图3 标题化合物对棉红蜘蛛杀虫活性的实验值与计算值的关系
Fig.3 Relationship between experimental and calculated insecticidal activities of title compounds to Tetranychus urticae
2.3 杀虫机理的分析
模型(4)~(7)显示影响标题化合物杀虫活性的共性因素是ELUMO与EHOMO的轨道能隙△E1。据福井谦一的前线分子轨道理论可知,分子在反应过程中优先起作用的是其前线分子轨道,如HOMO,NHOMO,LUMO和NLUMO等,它们对于分子的化学性质特别是其反应活性有着决定性作用。通常HOMO对其中的电子束缚较为松弛,具有电子给予体的性质;表现为EHOMO越高,其上电子越易失去而参加反应,具有较强的还原性。LUMO具有电子接受体的性质,表现为ELUMO越低,对电子的亲和力较强,越易接受电子而参加反应,呈现较强的氧化性。即EHOMO越高及ELUMO越低的苯并噻(噁)唑酮衍生物(NAB)具有较强的化学活泼性,相应表现出较强的生物活性即杀虫活性。对于EHOMO越高及ELUMO越低的标题化合物,其△E1越小,但杀虫活性SJ越高。即△E1与SJ为负相关关系,此与模型(4)~(7)中△E1前的系数均小于0是一致的。由此可见,NAB对蚕豆蚜、棉红蜘蛛的杀虫机理包含与其体内生物受体之间的电子转移反应。
由于蚕豆蚜、棉红蜘蛛的内部生理结构存在差别,对于同样的杀虫剂也会有应当差别的杀虫机理。模型(4)~(7)便显示出其间的区别:
(1) 影响NAB对蚕豆蚜杀虫活性的另一因素是亚胺氮原子上的电荷QN2。QN2反映BT与蚕豆蚜受体之间存在静电作用,其值越大,与受体的静电斥力越强,越难形成复合物,相应杀虫活性Sa越低。QN2中蕴含氢键作用,QN2越小,越易接受受体中电正性的氢形成氢键,导致杀虫活性增强。由此可见,QN2与其杀虫活性呈负相关,这与它们在模型(4)中的系数小于0相一致。
(2) 影响NAB对棉红蜘蛛杀虫活性的另一因素是次最低空轨道能ENLUMO。它与ELUMO具有类似的性质。通常认为分子的ENLUMO越小,越易接受电子而进入生物体内,导致其对棉红蜘蛛的毒性越大,即Su与ENLUMO应呈负相关。但模型(5)与(7)却显示两者正相关。对此可理解为:在△E1作用下,NAB已进入棉红蜘蛛体内并有电子进入NLUMO中;ENLUMO越高,在NLUMO中的电子越活泼,越易与受体生物大分子发生电子转移,致使其杀虫活性越大. 即Su与ENLUMO同向变化,这与模型(5)与(7)中其前系数大于零相一致。由此可见,NAB对棉红蜘蛛杀虫机理包含由△E1控制的电子转移及由ENLUMO控制的其他较复杂的电子反应。已有较多文献指出,电性参数ENLUMO进入模型,其机理中包含较复杂的电子反应[15]。此与本文结果一致。
苯甲酰基脲是典型的几丁质酶(Chitinase)抑制剂,其公认骨架结构为5部分组成(图4)。其中:Ar为芳基,R为烷基等,X和Y多为氧原子,亦可其中1个为硫原子。与图1比较可见,本文化合物基本骨架与其完全一致。因此,可以认为本文化合物亦可作为几丁质酶抑制剂。昆虫几丁质酶的氨基酸序列多由3个区域组成的多功能区:包括N2端的活性催化区,C端富含半胱氨酸的几丁质结合区,以及在这2个区域之间的被称为Linker的富含脯氨酸、谷氨酸、丝氨酸和苏氨酸的区域[16]。大多数已知的几丁质酶抑制剂与酶活性位点的结合是可逆的化学反应,即与活性部位的1个或多个色氨酸残基通过π-π键或其他的疏水键相互作用以及氢键结合。
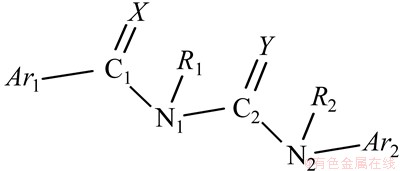
图4 苯甲酰基脲的基本结构
Fig.4 Basic structure of benzoylurea
对蚕豆蚜、棉红蜘蛛的杀虫活性最高的分子2的前沿分子轨道形状如图5所示,其形状均为π键轨道。相应分子的前线轨道成分与前沿电子密度见表3,其中:“AO”代表原子轨道;“FAOH”为最高占有分子轨道中主要原子轨道的系数;“EAH”为原子的最高占有轨道系数;“DAH”为原子的最高占有轨道的电子密度,以反映同一分子中不同原子的反应活性,其值越大,表明给出电子能力越强[17-18];“FAOL”为最低空分子轨道中主要原子轨道的系数,“EAL”代表原子的最低未占轨道系数,“DAL”为原子的最低未占轨道的电子密度,其值越大,接受电子能力越强,相应原子的反应活性在同一分子中较大[17-18]。应当说明的是:表3中的N13与N17分别对应图4中的N1与N2;C15与O16对应图4中的C2与Y;C19~C24对应图4中的Ar2;C7与O14对应图4中的C1与X。

图5 分子2的前沿分子轨道形状
Fig.5 Molecular orbital of molecule 2
表3 表1中分子2的前线轨道中的主要成分及分布
Table 3 Population of frontier molecular orbitals to some atoms of molecule 2 in Table 1

由图5及表3可见:分子2的前线分子轨道HOMO,LUMO等是π键轨道,且活性原子有N13,N17,C15,O16,C19~C24,C7和O14等。特别是N17与C19~C24构成活性催化区(因其电子密度很大,给出或接受电子能力很强), NAB与蚕豆蚜、棉红蜘蛛体内生物受体之间的电子转移反应很可能就发生在这些部位。因此,模型(4)~(7)的△E1及ENLUMO反映π-π键的相互作用。另外,QN2是揭示与酶受体间的氢键作用。综上所述,模型(4)~(7)所揭示的杀虫机理是与苯甲酰基脲类抑制几丁质酶的机理相一致。
2.4 结构修饰
QSAR研究的目的之一是根据模型设计分子并运用该模型去预测设计分子的活性,为新型的、高活性分子的合成提供理论依据。由前面的分析可知,降低△E1,QN2及提高ENLUMO的基团引入将有利于改善标题化合物对蚕豆蚜、棉红蜘蛛的杀虫活性。这也是进行分子结构改造,获得结构新颖,杀虫高效标题化合物的基本依据。据此,在图1中X=S及Y=Cl下,改变取代基R为CH3,NH2,Cl,Br和NO2,共设计19个化合物(表1中后19个化合物)。由表1可知,R为吸电子基团,使△E1及ENLUMO降低,相应地QN2升高,据模型(4)~(7)可知其杀虫活性增强。如第29至34号这6个化合物都含有强吸电子基团—硝基,它们对蚕豆蚜、棉红蜘蛛的杀虫活性都比较高。这是因为硝基的引入,能使△E1及ENLUMO显著降低(而QN2基变化不大)。特别是第29和33号这2个化合物对蚕豆蚜、棉红蜘蛛的杀虫活性均超过111.6%,表明这2个化合物可以在质量浓度低于500 mg/L时,仍能发挥良好的杀虫作用。这是因为硝基处于邻位时,△E1更小。当然,修饰后的这些化合物的杀虫活性有待生物活性试验予以验证。如能被证实,第29和33号这2个化合物有望开发为新型高效杀虫剂。
3 结论
(1) 采用考虑电子相关作用的DFT-B3LYP方法,在较高基组6-31G水平下获得34种N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺化合物的量化结构参数,它们对这些分子的结构差异具有良好的区分能力。只要保留足够的有效数字,量化参数就能实现分子结构的数值化表征。
(2) 利用最佳变量子集回归方法,建立上述量化参数对蚕豆蚜,棉红蜘蛛的杀虫活性SJ的二元数学模型(4)~(5),经R2,Radj2,F,Rcv2,VIF,AIC和FIT等质量指标检验,所建模型不仅显示高度相关,而且呈现良好的稳健性及预测能力,并经455个训练集与测试集予以验证。
(3) N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺分子对蚕豆蚜,棉红蜘蛛的杀虫机理是与苯甲酰基脲类抑制几丁质酶的机理一致。降低△E1,QN2及提高ENLUMO的基团引入将有利于改善标题化合物对蚕豆蚜、棉红蜘蛛的杀虫活性。由于强吸电子基团—硝基的引入,能使△E1及ENLUMO显著降低,有利于其与蚕豆蚜、棉红蜘蛛体内生物受体间的电子转移,致使其杀虫活性大幅度提高。
参考文献:
[1] 王伟, 张国平, 宋宝安, 等. O,O′-二烷基-α-(取代苯并噻唑-2-基)氨基-(取代苯基甲基)膦酸酯的合成与抗烟草花叶病毒活性[J]. 有机化学, 2007, 27(2): 279-284.
WANG Wei, ZHANG Guoping, SONG Baoan, et al. Synthesis and anti-tobacco mosaic virus activity of O,O′-dialkyl-α- (substituted benzothiazol-2-yl)amino-(substituted phenylmethyl) phosphonate[J]. Chin J Org Chem, 2007, 27(2): 279-284.
[2] 翁建全, 刘幸海, 张国富, 等. 过渡金属催化下N-取代苯基-苯并噻(噁)唑酮-3-甲酰胺的合成及其生物活性[J]. 有机化学, 2011, 31(3): 374-378.
WENG Jianquan, LIU Xinghai, ZHANG Guofu, et al. Transition-metal-catalyzed synthesis and biological activities of N-arylaminocarbonyl-2-benzothiazolinone/benzoxazolone[J]. Chin J Org Chem, 2011, 31(3): 374-378.
[3] CAO Dongsheng, LIANG Yizeng, XU Qingsong, et al. A new strategy of outlier detection for QSAR/QSPR[J]. Journal of Computational Chemistry, 2010, 31(3): 592-602.
[4] HUANG Xin, XU Qingsong, LIANG Yizeng. PLS regression based on sure independence screening for multivariate calibration[J]. Analytical Methods, 2012, 4(9): 2815-2821.
[5] 冯长君. 手性有机酸保留指数的手性指数及原子类型电拓扑指数模型[J]. 物理化学学报, 2010, 26(1): 193-198.
FENG Changjun. Mathematical model for retention indices, chiral index and electrotopological state indices for atom types of chiral organic acids[J]. Acta Physico-Chimica Sinica, 2010, 26(1): 193-198.
[6] 冯长君. 3-取代硫基-5-(2-羟基苯基)-4H-1,2,4-三唑类化合物抑菌活性的定量构效关系和结构修饰的理论研究[J]. 化学学报, 2012, 70(4): 512-518.
FENG Changjun. Theoretical studies on quantitative structure- activity relationship and structural modification for 3-substituted sulfur-5-(2-hydroxyphenyl)-4H-1,2,4-triazole compounds[J]. Acta Chimica Sinica, 2012, 70(4): 512-518.
[7] WEI Dongbin, ZHANG Aiqian, WU Chunde, et al. Progressive study and robustness test of QSAR model based on quantum chemical parameters for predicting BCF of selected polychlorinated organic compounds[J]. Chemosphere, 2001, 44(6): 1421-1428.
[8] 陈建华, 王进明, 龙贤灏, 等. 硫化铜矿物电子结构的第一性原理研究[J]. 中南大学学报: 自然科学版, 2011, 42(12): 3612-3617.
CHEN Jianhua, WANG Jinming, LONG Xianhao, et al. First-principle theory on electronic structure of copper sulfides[J]. Journal of Central South University: Science and Technology, 2011, 42(12): 3612-3617.
[9] 唐新村, 陈静波, 贾殿赠, 等. 氮川三乙酸的脱水反应机理[J]. 中南大学学报: 自然科学版, 2010, 41(2): 446-449.
TANG Xincun, CHEN Jingbo, JIA Dianzeng, et al. Process of dehydration of nitrilotriacetic acid[J]. Journal of Central South University: Science and Technology, 2010, 41(2): 446-449.
[10] 谢湖均, 雷群芳, 胡晓环, 等. 1-取代-2-氨基苯并咪唑衍生物的理论计算及QSAR研究[J]. 化学学报, 2011, 69(4): 399-404.
XIE Hujun, LEI Qunfang, HU Xiaohuan, et al. Theoretical calculations on 1-Substituted-2-amino-benzimidazole derivatives and studies on quantitative structure-activity relationship (QSAR)[J]. Acta Chimica Sinica, 2011, 69(4): 399-404.
[11] Urra L S, Gonza′lez M P, Teijeira M. 2D-autocorrelation descriptors for predicting cytotoxicity of naphthoquinone ester derivatives against oral human epidermoid carcinoma[J]. Bioorganic & Medicinal Chemistry, 2007, 15(10): 3565-3571.
[12] Urra L S, Gonza′lez M P, Teijeira M. QSAR studies about cytotoxicity of benzophenazines with dual inhibition toward both topoisomerases Ⅰ and Ⅱ: 3D-MoRSE descriptors and statistical considerations about variable selection[J]. Bioorganic & Medicinal Chemistry, 2006, 14(21): 7347-7358.
[13] 刘东, 章文军, 许禄. 手性羟酸和氨基酸类化合物的构效关系研究[J]. 化学学报, 2009, 67(2): 145-150.
LIU Dong, ZHANG Wenjun, XU Lu. Quantitative structure- activity/property relationships for chiral hydroxyl acids and amino acids[J]. Acta Chimica Sinica, 2009, 67(2): 145-150.
[14] Yao S W, Lopes V H C, Fernández F, et al. Synthesis and QSAR study of the anticancer activity of some novel indane carbocyclic nucleosides[J]. Bioorg Med Chem, 2003, 11(23): 4999-5006.
[15] 籍国东, 袁星, 赵元慧, 等. 应用次最低空轨道能研究硝基芳烃的生物活性[J]. 环境科学, 1999, 20(2): 68-70.
JI Guodong, YUAN Xing, ZHAO Yuanhui, et al. The study on biological activities of nitroaromatic compound using quantum chemistry descriptor of next lowest unoccupied molecular orbital[J]. Environmental Science, 1999, 20(2): 68-70.
[16] Arakane Y, ZHU Qingsong, Matsumiy M, et al. Properties of catalytic, linker and chitin-binding domains of insect chitinase[J]. Insect Biochem Mol Biol, 2003, 33(6): 631-648.
[17] 刘峥, 张小鸽, 袁帅, 等. 吡啶-4-甲醛缩氨基酸席夫碱及其配合物的抑菌性能和量子化学研究[J]. 计算机与应用化学, 2012, 29(6): 674-678.
LIU Zheng, ZHANG Xiaoge, YUAN Shuai, et al. Antibacterial activity and quantum chemistry of pyridine-4-formaldehyde- amino-acid and their metal complexes[J]. Computers and Applied Chemistry, 2012, 29(6): 674-678.
[18] 刘志国, 赵学辉, 谢志美. 1-(2,3,4-三羟基苯亚甲基)-4-羟基氨基脲结构与性能的DFT研究[J]. 湖南城市学院学报: 自然科学版, 2004, 13(1): 57-60.
LIU Zhiguo, ZHAO Xuehui, XIE Zhimei. Ab initio DFT on the structure-antitumor activity relationship of 1-(2,3,4-trihydroxy- henzylidene)-4-hydroxysemi-carbazide[J]. J Hunan City University: Natural Science, 2004, 13(1): 57-60.
(编辑 邓履翔)
收稿日期:2012-11-04;修回日期:2013-03-11
基金项目:国家自然科学基金资助项目(21075138);环境模拟与污染控制国家重点联合实验室开放基金资助项目(13K02ESPCT);徐州市科技局基金资助项目(XZZD1104);徐州市贾汪区科技局基金资助项目(XM10A05)
通信作者:冯长君(1954-),男,江苏徐州人,教授,从事量化计算及药物构效关系等研究;电话:0516-85608039;E-mail: fengcjxznu@xznu.edu.cn

