Au3Cu-亚格子系统的合金基因Gibbs能配分函数和平衡全息网络相图
来源期刊:中国有色金属学报(英文版)2015年第1期
论文作者:谢佑卿 聂耀庄 李小波 彭红建 刘心笔
文章页码:211 - 240
关键词:Au3Cu化合物;Au3Cu-型亚格子系统;合金基因Gibbs能配分函数;平衡全息网络相图;系统金属材料科学
Key words:Au3Cu compound; Au3Cu-type sublattice system; alloy gene Gibbs energy partition function; equilibrium holographic network phase diagrams; systematic metal materials science
摘 要:以Au3Cu-亚格子系统为例,介绍了3项发现:第一,至今阻碍金属材料科学进步的第四大障碍是研究者们尚未认识到一个真正的合金相Gibbs能函数应由合金基因序列和它们自己的Gibbs能级序列构建的Gibbs能配分函数导出。第二,建立合金基因Gibbs能配分函数的六条规则,特别证明了计算合金组态熵的简并因子中结构单元占居Gibbs能级的概率应按照组元占居格点的概率方式简并;第三,以前研究者从未预料到的主要特征:具有一条没有有序相和无序相共存区的单相相界线;相界线顶点成分和温度远偏离Au3Cu化合物临界点的成分和温度;在0 K时,Gibbs能随成分变化曲线上的最低点成分远偏离Au3Cu化合物的成分;Au3Cu-型长程有序合金的理论极限成分范围由第一跳变有序度决定。
Abstract: Taking Au3Cu-type sublattice system as an example, three discoveries have been presented. First, the fourth barrier to hinder the progress of metal materials science is that today’s researchers do not understand that the Gibbs energy function of an alloy phase should be derived from Gibbs energy partition function constructed of alloy gene sequence and their Gibbs energy sequence. Second, the six rules for establishing alloy gene Gibbs energy partition function have been discovered, and it has been specially proved that the probabilities of structure units occupied at the Gibbs energy levels in the degeneracy factor for calculating configuration entropy should be degenerated as ones of component atoms occupied at the lattice points. Third, the main characteristics unexpected by today’s researchers are as follows. There exists a single-phase boundary curve without two-phase region coexisting by the ordered and disordered phases. The composition and temperature of the top point on the phase-boundary curve are far away from those of the critical point of the Au3Cu compound; At 0 K, the composition of the lowest point on the composition-dependent Gibbs energy curve is notably deviated from that of the Au3Cu compounds. The theoretical limit composition range of long range ordered Au3Cu-type alloys is determined by the first jumping order degree.
Trans. Nonferrous Met. Soc. China 25(2015) 211-240
Youq-qing XIE1,2,3, Yao-zhuang NIE4, Xiao-bo LI5, Hong-jian PENG6, Xin-bi LIU1,2,3
1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Powder Metallurgy Research Institute, Central South University, Changsha 410083, China;
3. State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China;
4. School of Physics and Electronics, Central South University, Changsha 410083, China;
5. School of Materials Science and Engineering, Xiangtan University, Xiangtan 411105, China;
6. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
Received 17 November 2014; accepted 8 January 2015
Abstract: Taking Au3Cu-type sublattice system as an example, three discoveries have been presented. First, the fourth barrier to hinder the progress of metal materials science is that today’s researchers do not understand that the Gibbs energy function of an alloy phase should be derived from Gibbs energy partition function constructed of alloy gene sequence and their Gibbs energy sequence. Second, the six rules for establishing alloy gene Gibbs energy partition function have been discovered, and it has been specially proved that the probabilities of structure units occupied at the Gibbs energy levels in the degeneracy factor for calculating configuration entropy should be degenerated as ones of component atoms occupied at the lattice points. Third, the main characteristics unexpected by today’s researchers are as follows. There exists a single-phase boundary curve without two-phase region coexisting by the ordered and disordered phases. The composition and temperature of the top point on the phase-boundary curve are far away from those of the critical point of the Au3Cu compound; At 0 K, the composition of the lowest point on the composition-dependent Gibbs energy curve is notably deviated from that of the Au3Cu compounds. The theoretical limit composition range of long range ordered Au3Cu-type alloys is determined by the first jumping order degree.
Key words: Au3Cu compound; Au3Cu-type sublattice system; alloy gene Gibbs energy partition function; equilibrium holographic network phase diagrams; systematic metal materials science
1 Introduction
In the first paper of a series articles for the FCC-based lattice Au-Cu system [1], we presented four philosophic propositions of system sciences. The first proposition is that “A diversity of structures of a system is attributed to the combination and arrangement of structural units in the basic structure unit sequences”. We delivered an extensive definition of the gene sequence: the gene sequence is a basic structure unit sequence carrying a set of transmission information about structures and properties for determining the diversity of structures and properties of a system, which may be a biologic or non-biologic system. Therefore, to seek alloy gene (AG) sequences is the first important task for establishing the systematic metal materials science (SMMS) framework [2,3], and also the first barrier to hinder the progress of metal materials science. The AG sequences are the  - and
- and  -central characteristic atom sequences in the
-central characteristic atom sequences in the 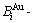 and
and  -basic coordination cluster sequences for the FCC-based lattice Au-Cu system. Their potential energies (
-basic coordination cluster sequences for the FCC-based lattice Au-Cu system. Their potential energies (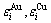 ) and volumes (
) and volumes ( ) sequences were obtained by the separated theory of potential energies and volumes of characteristic atoms. The electronic structures, physical properties and complete thermodynamic properties were obtained by valence bond theory and thermodynamics of characteristic crystals, respectively. These data were deposited into the AG-holographic information database.
) sequences were obtained by the separated theory of potential energies and volumes of characteristic atoms. The electronic structures, physical properties and complete thermodynamic properties were obtained by valence bond theory and thermodynamics of characteristic crystals, respectively. These data were deposited into the AG-holographic information database.
The second proposition is that “The diversity of properties of a system is attributed to contents and transmission mode of the information of basic structure unit sequences”. Therefore, to seek transmission mode of AG-information is the second task for establishing the SMMS framework, and also the second barrier to hinder the progress of metal materials science. The transmission mode of AG-information is described by AG-Gibbs energy 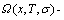 partition function, which consists of the AG-characteristic Gibbs energy transmission
partition function, which consists of the AG-characteristic Gibbs energy transmission  partition function and AG-arranging (AGA)
partition function and AG-arranging (AGA)  degeneracy function.
degeneracy function.
“The complexity and entirety of a complex system are attributed to the multilevel of the structures and properties and to the various correlativities between different structural levels, between different properties and between structure and property”. It is the third philosophic proposition of system sciences. The SMMS framework should be divided into three levels: 1) AG-theory to establish AG-holographic information database, which includes separated theory of potential energies and volumes of characteristic atoms (alloy genes), valence bond theory and thermodynamics of characteristic crystals [4-7]. 2) AGA-theory of alloy phases to establish AGA-holographic network information database, which includes AGA- crystallographics [8], AGA-valence bond theory [9-12] and AGA-thermodynamics of alloy phases [1,13,14]. 3) Alloy phase arranging (APA) theory of organizations, which includes APA-thermodynamics and APA-structure theory of alloy systems. “Properties are determined by structures, properties should be suitable for environments and environments change structures”. The man’s knowledge of relationships of structures, properties and temperature for alloys has been changed from single causality to systematic correlativity, due to the discoveries of alloy gene sequences and their information transmission modes, as well as establishments of the SMMS framework, holographic alloy positioning (HAP) system, and equilibrium holographic network phase (EHNP) diagrams.
In the second paper [13], we proposed the fourth proposition: “The system has not only the ability to keep structure stabilization against a changing environment, but also a mechanism to change structure for suiting variation in environments”. Therefore, to seek atom movement mechanism is the third important task for establishing kinetics of alloy phase transformations, and also the third barrier to hinder the progress of metal materials science. Taking experimental path on disordering AuCuI ( ) composed of
) composed of 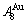 and
and  stem alloy genes as an example, we presented three discoveries: 1) The ability of the AuCuI() to keep structure stabilization against changing temperature is attributed to that the
stem alloy genes as an example, we presented three discoveries: 1) The ability of the AuCuI() to keep structure stabilization against changing temperature is attributed to that the 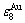 and
and  potential well depths greatly surpass their vibration energies, which leads to experimental path in subequilibrium state. 2) A new atom movement mechanism of AuCuI
potential well depths greatly surpass their vibration energies, which leads to experimental path in subequilibrium state. 2) A new atom movement mechanism of AuCuI to change structure for suiting variation in temperature is the “resonance activating-synchro alternating” of alloy genes, and it leads to heterogeneous nucleation and successive subequilibrium transitions. 3) There exists jumping order degree, which leads to the existence of jumping Tj-temperature and an unexpected so-called “retro-effect” about jumping temperature retrograde shift to lower temperatures upon the increasing heating rate [15-17].
to change structure for suiting variation in temperature is the “resonance activating-synchro alternating” of alloy genes, and it leads to heterogeneous nucleation and successive subequilibrium transitions. 3) There exists jumping order degree, which leads to the existence of jumping Tj-temperature and an unexpected so-called “retro-effect” about jumping temperature retrograde shift to lower temperatures upon the increasing heating rate [15-17].
In the third paper [14], taking AuCu3-type sublattice system as an example, we presented three discoveries: 1) The third barrier to hinder progress of metal materials science is that researchers have got used to recognizing experimental phenomena of alloy phase transitions during extremely slow variation in temperature by equilibrium thinking mode and then to take miss knowledge of experimental phenomena as selected information for establishing so-called Gibbs energy function and so-called equilibrium phase diagram. 2) The EHNP-diagrams of AuCu3-type sublattice system may be used to describe systematic correlativity of the composition-temperature-dependent alloy gene arranging structures and complete thermodynamic properties, and to be a standard for studying experimental subequilibrium order-disorder transition. 3) The main characteristics of these EHNP-diagrams have never been expected by today’s researchers.
In the present work, taking Au3Cu-type sublattice system as an example, we presented three discoveries: 1) The fourth barrier to hinder the progress of metal materials sciences is that today’s researchers do not understand that a real Gibbs energy function of an alloy phase should be derived from corresponding AG-Gibbs energy partition function constructed of AG-sequence and their Gibbs energy sequence. 2) To establish AG-Gibbs energy partition function should obey six rules, which can make us to understand problematic essentials of the currently used alloy solution models in quantum mechanical abinitio calculations of (QMAC)- thermodynamics and calculation of phase diagrams (CALPHAD)-thermodynamics. 3) The main characteristics of equilibrium holographic network phase diagrams of the Au3Cu-type sublattice system have never been expected by today’s researchers, which may be used to explain some wondering experimental phenomena.
2 Rules for establishing AG-Gibbs energy partition function
In spite of a long history of statistical thermodynamics [18-22], there is none of the currently used so-called Gibbs energy functions of alloy phases in QMAC- and CALPHAD-thermodynamics to be a real Gibbs energy function derived from the real Gibbs energy partition function. It shows that today’s researchers do not understand that a real Gibbs energy function should be derived from the real Gibbs energy partition function constructed of the basic structure unit sequences and their Gibbs energy sequences. It is the fourth barrier to hinder the progress of the metal materials science, because some problems existed in these so-called Gibbs energy functions can not be easily overcome.
Based on the AG-Gibbs energy sequences and AGA-model, the AG-Gibbs energy partition function of the FCC-based lattice Au-Cu system has been established, which is used to describe the systematic correlativity of the AG-Gibbs energy levels (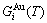 ,
,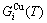 ), AG-probabilities (
), AG-probabilities ( ,
,  ) occupied at the AG-Gibbs energy levels and degeneracy
) occupied at the AG-Gibbs energy levels and degeneracy 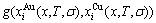 factor of AG-probabilities as functions of the composition (x), temperature (T) and order degree (
factor of AG-probabilities as functions of the composition (x), temperature (T) and order degree (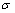 ):
):
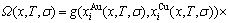
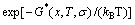 (1)
(1)
where kB is the Boltzmann’s constant, and 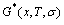 is the characteristic Gibbs energy function of the alloy phase, which may be obtained by the transmission law of the AG-Gibbs energies:
is the characteristic Gibbs energy function of the alloy phase, which may be obtained by the transmission law of the AG-Gibbs energies:

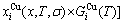 (2)
(2)
The Gibbs energy function of the alloy phase may be derived from the 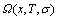 -function:
-function:

 (3)
(3)
The configurational entropy 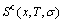 -function is obtained from the degeneracy g-factor:
-function is obtained from the degeneracy g-factor:
 (4)
(4)
It should be pointed out that these functions may be suitable for Au3Cu-, AuCu- and AuCu3-type sublattice systems (σ>0), as well as disordered alloy phase (σ=0). However, their ordering definitions are different, of which the details can be seen from Ref. [23]. We have discovered six rules for establishing AG-Gibbs energy partition function, which may reveal problematic essentials of the currently used alloy solution models in the QMAC-thermodynamics [24] and the CALPHAD- thermodynamics [25-28].
1) “The structure units should be suited to alloy phase-structure level”. An alloy phase is an alloy gene arranging (AGA) solution. The AG-sequences, AG-pair sequences and AG-cluster sequences may be used as structure unit sequences to establish Gibbs energy partition function, rather than “constituent atoms”, “constituent atom pairs” and “constituent atom clusters”. The AG-sequences carry structure information about coordinative configurations of theirs coordination clusters to describe diversity of AGA-phase structures and valences electron structures to explain essence of diversity of AGA-phase structures. However, the constituent units are not complete sequences with less information about only structures and properties of pure elements (see Appendix A.1).
2) “Energy level sequences should be AG-Gibbs energy sequences”. The objects of the AG-Gibbs energy partition function of an alloy phase are to derive the AGA-Gibbs energy function and to obtain the equilibrium order-disorder transition  -path with minimum Gibbs energy of alloys as functions of composition, temperature and order degree, then to obtain equilibrium holographic network path (EHNP) charts of alloys and to establish EHNP-diagrams of alloy system, because each Gibbs energy level may be divided into two parts: a temperature-independent contribution of chemical potential energy(E(0)), of which the variation with temperature has been accounted in the attaching vibration energy, and a temperature- independent contribution of generalized vibration free energy
-path with minimum Gibbs energy of alloys as functions of composition, temperature and order degree, then to obtain equilibrium holographic network path (EHNP) charts of alloys and to establish EHNP-diagrams of alloy system, because each Gibbs energy level may be divided into two parts: a temperature-independent contribution of chemical potential energy(E(0)), of which the variation with temperature has been accounted in the attaching vibration energy, and a temperature- independent contribution of generalized vibration free energy  level, which includes the generalized vibration energy
level, which includes the generalized vibration energy  and generalized vibration entropy
and generalized vibration entropy  . The includes Debye vibration energy
. The includes Debye vibration energy  and attaching vibration energy
and attaching vibration energy 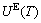 . The includes Debye vibration entropy (
. The includes Debye vibration entropy ( ) and attaching vibration entropy (
) and attaching vibration entropy ( ). The
). The  and include contributions of electron excitation, hole energy of formation, volume expansion work and variation in potential energy with temperature. If the generalized vibration free energy is neglected, the AG-Gibbs energy partition function becomes the AG-“potential energy” partition function [20], from which only the AGA-“potential free energy” function of alloy phases can be derived, then to obtain the so-called equilibrium order-disorder transition Emin-T path with minimum “potential free energy” of the alloy. However, it may be proved that this alloy is not in equilibrium state, and it is minimal “potential energy Emin-path is higher than the minimal Gibbs energy Gmin-path, because the negative value contributions of the AG-generalized vibration entropy energies surpass positive value contributions of the AG-generalized vibration entropy energies to AG-Gibbs energies (see Appendixes A.2 and A.3). It is also demonstrated that the AG-probabilities occupied at the AG-potential energy levels in the equilibrium state should be calculated by the minimal Gibbs energy path method.
and include contributions of electron excitation, hole energy of formation, volume expansion work and variation in potential energy with temperature. If the generalized vibration free energy is neglected, the AG-Gibbs energy partition function becomes the AG-“potential energy” partition function [20], from which only the AGA-“potential free energy” function of alloy phases can be derived, then to obtain the so-called equilibrium order-disorder transition Emin-T path with minimum “potential free energy” of the alloy. However, it may be proved that this alloy is not in equilibrium state, and it is minimal “potential energy Emin-path is higher than the minimal Gibbs energy Gmin-path, because the negative value contributions of the AG-generalized vibration entropy energies surpass positive value contributions of the AG-generalized vibration entropy energies to AG-Gibbs energies (see Appendixes A.2 and A.3). It is also demonstrated that the AG-probabilities occupied at the AG-potential energy levels in the equilibrium state should be calculated by the minimal Gibbs energy path method.
In the QMAC-thermodynamics, the effective cluster interaction parameters are obtained by fitting the  -enthalpy of formation, i.e.,
-enthalpy of formation, i.e.,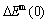 -potential energy of formation, calculated from the first-principles at electron theory T=0 K, here, the generalized vibration free energy is neglected [29]. Consequently, only the potential free energy function can be obtained by this way.
-potential energy of formation, calculated from the first-principles at electron theory T=0 K, here, the generalized vibration free energy is neglected [29]. Consequently, only the potential free energy function can be obtained by this way.
In the CALPHAD-thermodynamics, the Gibbs energies of the pure elements are taken from the compilation of DINSDALE [30], and the excess Gibbs energies of the ordered and disordered alloy phases are modeled with a Gibbs energy expression in the compound energy formalism or in the form of a Redlich- Kister series, that contains a number of unknown parameters. These parameters may be adjusted until the Gibbs energy expression corresponding to the chosen model is capable of representing the selected information well [31]. Consequently, this so-called Gibbs energy function is questionable, based on the misunderstanding of that the middle jumping Tj-temperature is erroneously considered as the terminal Tc-critical temperature of order-disorder equilibrium transition.
3) “The Gibbs energy levels should be consistent with their structure units”. In the Au-Cu system, the Au(1-x)Cux alloy may be defined as the “alloy gene arranging solution”, where the  and
and  alloy genes are regarded as the “structure units”, which should be matched by “AG-Gibbs energy levels”. The alloy may be also defined as the “alloy gene pair arranging solution”, where the
alloy genes are regarded as the “structure units”, which should be matched by “AG-Gibbs energy levels”. The alloy may be also defined as the “alloy gene pair arranging solution”, where the 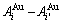 ,
,  and
and 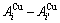 alloy gene pairs are regarded as the “structure units”, which should be matched by “alloy gene pair Gibbs energy levels”. The alloy may be also defined as the “alloy gene cluster arranging solution”, where the “alloy gene clusters” are regarded as the “structure units”, which should be matched by “alloy gene cluster Gibbs energy levels”. If the alloy is defined as the Au- and Cu-atom solution mixed by pure Au and Cu metals, where the Au- and Cu-atoms are regarded as the “constituent units”. It is well known that after mixing Au- and Cu-pure elements, their energy states are split, so, there exist uncertain Gibbs energy levels to match them. Therefore, the QMAC and CALPHAD so-called Gibbs energy functions contain a number of unknown parameters, that may be understood from Appendixes A.2 and A.3.
alloy gene pairs are regarded as the “structure units”, which should be matched by “alloy gene pair Gibbs energy levels”. The alloy may be also defined as the “alloy gene cluster arranging solution”, where the “alloy gene clusters” are regarded as the “structure units”, which should be matched by “alloy gene cluster Gibbs energy levels”. If the alloy is defined as the Au- and Cu-atom solution mixed by pure Au and Cu metals, where the Au- and Cu-atoms are regarded as the “constituent units”. It is well known that after mixing Au- and Cu-pure elements, their energy states are split, so, there exist uncertain Gibbs energy levels to match them. Therefore, the QMAC and CALPHAD so-called Gibbs energy functions contain a number of unknown parameters, that may be understood from Appendixes A.2 and A.3.
4) “The used structure units should be the smallest ones”. As compared with AG-pair and AG-cluster, the alloy gene is the smallest. The AG-Gibbs energy partition function and derived AGA-Gibbs energy function are simplest. It may be proved that if the AG-sequence, AG-pair sequence and AG-cluster sequence are the respectively complete sequences, the total Gibbs energies, characteristic Gibbs energies and configurational entropies calculated by them are identical. So, we should adopt the AG-sequence as the structure unit sequence, rather than AG-pair and AG-pair sequences for simplified calculation (see Appendixes A.2 and A.3). In the QMAC- and CALPHAD- thermodynamics, the atomic pairs and atomic clusters are the incomplete “constituent” units. The total Gibbs energies and configurational entropies calculated by different “constituent units” are different, respectively.
5) “A set of structure unit sequences with corresponding Gibbs energy sequences should be suitable for all phases in a based lattice system”. The Au-Cu system with fcc-based lattice possesses a set of and  sequences with a set of
sequences with a set of  and
and  sequences and other properties sequences, which may be suitable for describing Au3Cu-, AuCu- and AuCu3-type ordered phases, as well as disordered phase. Therefore, their Gibbs energy functions have no any adjustable parameters. The Al-Ni system with FCC- and BCC-based lattices possesses two sets of
sequences and other properties sequences, which may be suitable for describing Au3Cu-, AuCu- and AuCu3-type ordered phases, as well as disordered phase. Therefore, their Gibbs energy functions have no any adjustable parameters. The Al-Ni system with FCC- and BCC-based lattices possesses two sets of  and
and  sequences. The Al-Ti system with FCC-, HCP and BCC-based lattices possesses three sets of and
sequences. The Al-Ti system with FCC-, HCP and BCC-based lattices possesses three sets of and 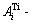 sequences.
sequences.
6) “The AG-probabilities occupied at the Gibbs energy levels should be degenerated by the probabilities of component atoms occupied at the sublattice points in the degeneracy function’’. In the AG-Gibbs energy partition function, the characteristic Gibbs energy function and degeneracy 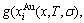
 function are interrelated, because both they are associated with AG-propabilities occupied at the Gibbs energy levels. The relation of molar configurational entropy to degeneracy function is described by Boltzman’s function (see Appendix A.4). Therefore,
function are interrelated, because both they are associated with AG-propabilities occupied at the Gibbs energy levels. The relation of molar configurational entropy to degeneracy function is described by Boltzman’s function (see Appendix A.4). Therefore,  function associated with the function is not independent contributor to Gibbs energy of the alloy phase.
function associated with the function is not independent contributor to Gibbs energy of the alloy phase.
In the fourth rule, it has been pointed out that the Gibbs energy, characteristic Gibbs energy and configurational entropies calculated respectively by AG-, AG-pair and AG-cluster sequences should be identical, if they are complete sequences and matched by their Gibbs energy sequences. It means that the calculated configurational entropies are independent on the sizes of structure units. We have also calculated configurational entropies by three degeneracy  -functions. It has been proved that the AG-probabilities occupied at the AG-Gibbs energy levels in the degeneracy function should be degenerated by those of the constituent units occupied at the sublattice points, i.e., Bragg-William model [32,33] (see Appendix A.4), because the AG-generalized vibration entropy energy
-functions. It has been proved that the AG-probabilities occupied at the AG-Gibbs energy levels in the degeneracy function should be degenerated by those of the constituent units occupied at the sublattice points, i.e., Bragg-William model [32,33] (see Appendix A.4), because the AG-generalized vibration entropy energy 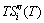 has been accounted in the AG-Gibbs energy level (see Appendix A.2) .
has been accounted in the AG-Gibbs energy level (see Appendix A.2) .
In the QMAC- and CAPPHAD-thermodynamics, the configurational entropy is regarded as independent contributor to Gibbs energy and dependent on the sizes of constituent units, because the atomic pairs and atomic clusters are the incomplete constituent units.
3 EHNP-diagrams of Au3Cu-type sublattice system
3.1 Steps for establishing EHNP-diagrams
An equilibrium order/disorder transition for a given composition alloy is defined as that “the AG-Gibbs energy levels ( ,
,  ) and AG-probabilities (
) and AG-probabilities ( ,
, 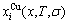 ) occupied at the - and -energy levels can respond immediately and change synchronously with each small variation in temperature and proceed along the minimal Gibbs energy path, supposing that there is no obstacle to atom movement”. The man’s knowledge of relationships of structures, properties and temperature for alloys has been changed from single causality to systematic correlativity, due to the discovery of alloy gene
) occupied at the - and -energy levels can respond immediately and change synchronously with each small variation in temperature and proceed along the minimal Gibbs energy path, supposing that there is no obstacle to atom movement”. The man’s knowledge of relationships of structures, properties and temperature for alloys has been changed from single causality to systematic correlativity, due to the discovery of alloy gene  and sequences and establishment of the AG-Gibbs energy partition function. The systematic correlativity of the Au3Cu-type sublattice system may be described by a set of EHNP diagrams, which are obtained by following steps.
and sequences and establishment of the AG-Gibbs energy partition function. The systematic correlativity of the Au3Cu-type sublattice system may be described by a set of EHNP diagrams, which are obtained by following steps.
1) According to the and level sequences, mixed Gibbs energy 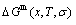 -function and essential definition of equilibrium order-disorder transition, the systematic correlativity of the
-function and essential definition of equilibrium order-disorder transition, the systematic correlativity of the 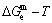 ,
, 
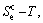
 and
and 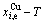 paths on equilibrium order-disorder transition as function of temperature for the stoichiometric Au3Cu-alloy are calculated by the minimal mixed Gibbs energy
paths on equilibrium order-disorder transition as function of temperature for the stoichiometric Au3Cu-alloy are calculated by the minimal mixed Gibbs energy 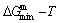 path method, which includes the iso-order degree Gibbs energy
path method, which includes the iso-order degree Gibbs energy 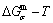 path method (Fig. 1(a)) and the isothermal Gibbs energy
path method (Fig. 1(a)) and the isothermal Gibbs energy  path method (Fig. 1(b)), using the calculated steps ΔT=1 K and Δσ=0.0001. According to the and paths, the EHNP charts of the stoichiometric Au3Cu-alloy are calculated and shown in Appendix B.
path method (Fig. 1(b)), using the calculated steps ΔT=1 K and Δσ=0.0001. According to the and paths, the EHNP charts of the stoichiometric Au3Cu-alloy are calculated and shown in Appendix B.
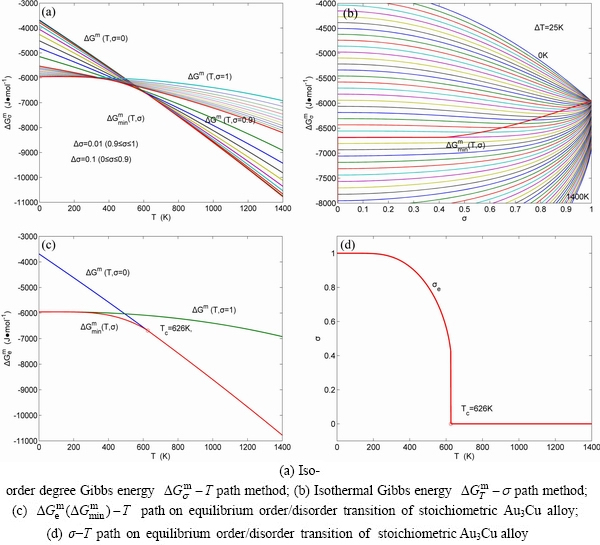
Fig. 1 Minimal mixed Gibbs energy 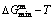 path on equilibrium order-disorder transition of stoichiometric Au3Cu alloy
path on equilibrium order-disorder transition of stoichiometric Au3Cu alloy
2) By analogy with the first step, the systematic correlativity data of the 
 ,
, 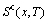 ,
, 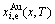 and
and  paths on equilibrium order-disorder transition as function of composition and temperature for all alloys of the Au3Cu-type sublattice system are calculated, using calculated steps Δx=0.5%, ΔT=1 K, Δσ=0.0001.
paths on equilibrium order-disorder transition as function of composition and temperature for all alloys of the Au3Cu-type sublattice system are calculated, using calculated steps Δx=0.5%, ΔT=1 K, Δσ=0.0001.
3) According to the systematic correlativity data of the  , , , and
, , , and 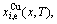 the three-dimensional
the three-dimensional 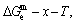
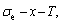
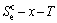 ,
,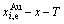 and
and  EHNP diagrams, as well as their two-dimensional
EHNP diagrams, as well as their two-dimensional 
 and
and 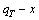 path network phase diagrams and configuration entropy EHNP diagrams are constructed and shown in Figs. 2-5.
path network phase diagrams and configuration entropy EHNP diagrams are constructed and shown in Figs. 2-5.
4) According to the first order thermodynamic properties in the AG-database and and EHNP diagrams, the EHNP diagrams of the first order thermodynamic properties of the Au3Cu-type sublattice system are calculated by their transmission laws, which may be also called as the additive law of extensive q-properties of characteristic crystals [1,34]. q denotes characteristic Gibbs energy (G*), enthalpy (H), potential energy (E), volume (V), generalized vibration free energy (Xv), generalized vibration energy (Uv), generalized vibration entropy (Sv).
5) The EHNP diagrams of the second order thermodynamic properties (mixed heat capacity and mixed volume expansion coefficient) are calculated [1,13].
6) The EHNP diagrams of the activities of the Au- and Cu-components are calculated [1,13].
7) The composition-temperature-dependent 
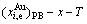 and
and  phase boundary (PB) curves are calculated by difference method of Gibbs energies between ordered and disordered phases (Appendix C)
phase boundary (PB) curves are calculated by difference method of Gibbs energies between ordered and disordered phases (Appendix C)
8) According to the and phase boundary curves, the other 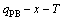 phase boundary curves are calculated.
phase boundary curves are calculated.
3.2 Mixed Gibbs energy EHNP diagrams
The mixed Gibbs energy EHNP diagrams include a three-dimensional  network phase diagram and three two-dimensional
network phase diagram and three two-dimensional  ,
,  and
and  path network phase diagrams (Fig. 2). In these diagrams, once one network point
path network phase diagrams (Fig. 2). In these diagrams, once one network point  has been clicked, the information about composition (x), temperature (T) and mixed Gibbs energy (
has been clicked, the information about composition (x), temperature (T) and mixed Gibbs energy ( ) may be readily obtained.
) may be readily obtained.
Fig. 2 Mixed Gibbs energy EHNP diagrams of Au3Cu-type sublattice system
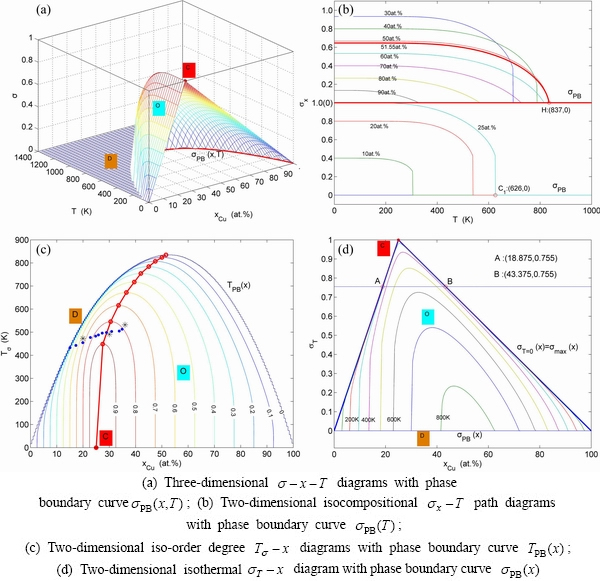
Fig. 3 Order degree EHNP diagrams of Au3Cu-type sublattice system
From Fig. 2(a), the following main understandings may be obtained. 1) There are ordered single-phase region with Gibbs energy network points (denoted by the symbol “O”) and disordered single-phase region with Gibbs energy network points (denoted by the symbol “D”). 2) The  -phase boundary (PB) curve with Gibbs energy network points is obtained by difference method of Gibbs energies between ordered and disordered phases. By this method, it has been proved that there is a single-phase boundary curve rather than a boundary two-phase region of ordered and disordered phases: 3) The equilibrium state of the stoichiometric Au3Cu compound, of which the alloy gene arranging (AGA)-molecular formula is the
-phase boundary (PB) curve with Gibbs energy network points is obtained by difference method of Gibbs energies between ordered and disordered phases. By this method, it has been proved that there is a single-phase boundary curve rather than a boundary two-phase region of ordered and disordered phases: 3) The equilibrium state of the stoichiometric Au3Cu compound, of which the alloy gene arranging (AGA)-molecular formula is the 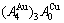 , exists only at the network C-point (
, exists only at the network C-point ( =25%, T=0 K, =-5955.72 J/mol).
=25%, T=0 K, =-5955.72 J/mol).
From Fig. 2(b), the following main understandings may be obtained. 1) The critical network point, i.e., phase boundary point, of the stoichiometric Au3Cu alloy, is located at the network C1-point (=25%,  =626 K, =-6685.96 J/mol). 2) The Au48.35Cu51. 65 alloy has the highest critical temperature in the Au3Cu-type sublattice system, it is located at the network H-point (=51.55%, Tc=837 K, =-10220.60 J/mol). 3) The equilibrium
=626 K, =-6685.96 J/mol). 2) The Au48.35Cu51. 65 alloy has the highest critical temperature in the Au3Cu-type sublattice system, it is located at the network H-point (=51.55%, Tc=837 K, =-10220.60 J/mol). 3) The equilibrium 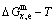 path for a given composition (x) alloy is the standard path for determining Gibbs energy hysteresis effect, i.e., superheated and undercooled driving Gibbs energies (
path for a given composition (x) alloy is the standard path for determining Gibbs energy hysteresis effect, i.e., superheated and undercooled driving Gibbs energies (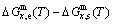 ) of experimental
) of experimental  path.
path.
From Fig. 2(c), we can know that the lowest network points of the iso-mixed Gibbs energy curves move from the network L-point (=43.40%, T=0 K, = =-6518.13 J/mol) to the network H-point (=51.55%, =837 K, =-10220.60 J/mol), that is unexpected by today’s researchers.
=-6518.13 J/mol) to the network H-point (=51.55%, =837 K, =-10220.60 J/mol), that is unexpected by today’s researchers.
From Fig. 2(d), we can know that the Au56.60Cu43.40 alloy with the lowest potential energy is located at the network L-point, which notably deviates from the C-point, that is also unexpected by today’s researchers. This diagram will be used to establish EHNP diagrams of the Au-Cu system together with isothermal  diagrams of the AuCu-type and AuCu3-type sublattice systems.
diagrams of the AuCu-type and AuCu3-type sublattice systems.
3.3 Order degree EHNP diagrams
The order degree EHNP diagrams include a three-dimensional  network phase diagram and three two-dimensional
network phase diagram and three two-dimensional  ,
,  and
and  path network phase diagrams (Fig. 3). In these diagrams, once one network point has been clicked, the information about composition (x), temperature (T) and order degree (σ), as well as the mixed Gibbs energy () may be readily obtained, because the order degree EHNP diagrams have been attached to the mixed Gibbs energy EHNP diagrams.
path network phase diagrams (Fig. 3). In these diagrams, once one network point has been clicked, the information about composition (x), temperature (T) and order degree (σ), as well as the mixed Gibbs energy () may be readily obtained, because the order degree EHNP diagrams have been attached to the mixed Gibbs energy EHNP diagrams.
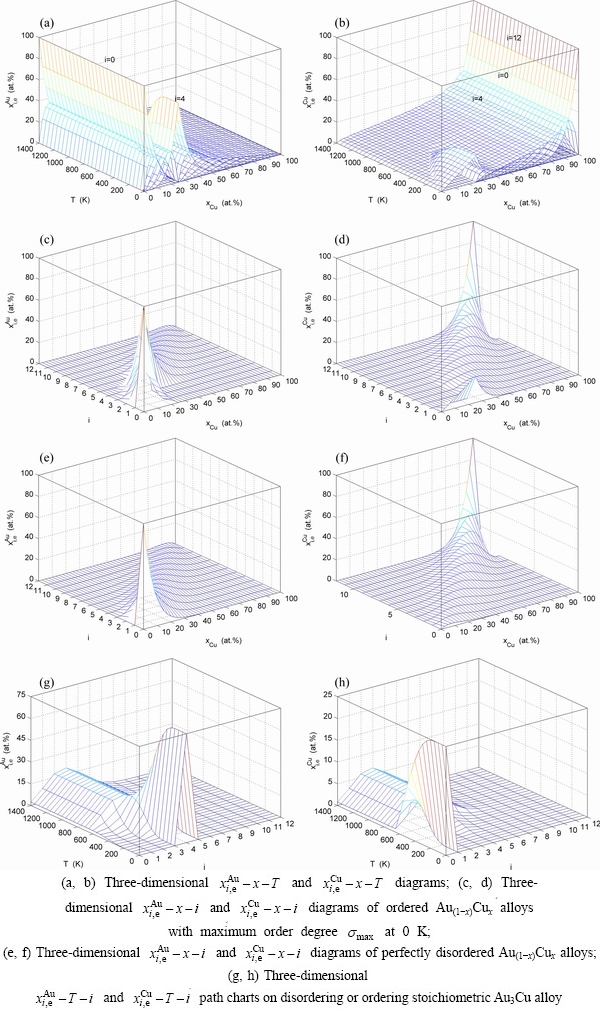
Fig. 4 AG-concentration EHNP diagrams
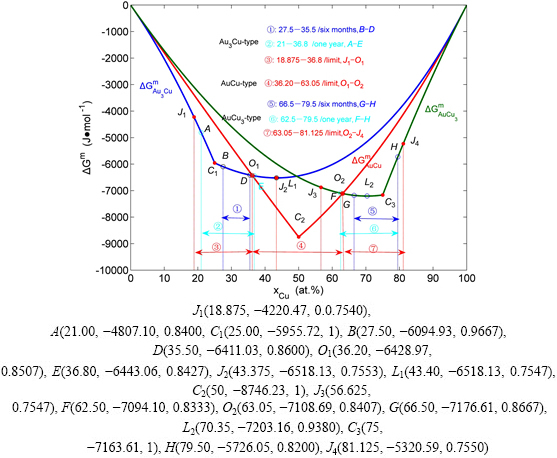
Fig. 5 Characteristics of 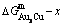 ,
, 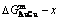 and
and  curves of Au3Cu-type, AuCu-type and AuCu3-type sublattice systems (Each point is described by composition, mixed Gibbs energy and order degree
curves of Au3Cu-type, AuCu-type and AuCu3-type sublattice systems (Each point is described by composition, mixed Gibbs energy and order degree
From Fig. 3(a), the following main understandings may be obtained. 1) There are ordered phase region with order degree network points (denoted by the symbol “O”, σ>0), disordered phase region (denoted by the symbol “D”, σ=0) and  -phase boundary (PB) curve (σ=0). 2) The equilibrium state of the compound exists only at the network C-point (=25%, T=0 K, σ=1).
-phase boundary (PB) curve (σ=0). 2) The equilibrium state of the compound exists only at the network C-point (=25%, T=0 K, σ=1).
From Fig. 3(b), we can know that: 1) The equilibrium 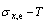 paths of alloys on the Au-rich side of the compound have great difference from thoes of alloys on the Cu-rich side of the compound. 2) The Au48.45Cu51.55 alloy in the single Au3Cu-type sublattice system has the highest critical temperature, and its network H-point (=51.55%, Tc=837 K, σ=0) deviates far from the network C1-point (=25%, Tc=626 K, σ=0) of stoichiometric Au3Cu alloy. However, their network points are respectively (=51.55%, σ=0.64) and (=25%, σ=1) at 0 K. 3) The equilibrium path for a given composition (x) alloy is the standard path for studying subequilibrium
paths of alloys on the Au-rich side of the compound have great difference from thoes of alloys on the Cu-rich side of the compound. 2) The Au48.45Cu51.55 alloy in the single Au3Cu-type sublattice system has the highest critical temperature, and its network H-point (=51.55%, Tc=837 K, σ=0) deviates far from the network C1-point (=25%, Tc=626 K, σ=0) of stoichiometric Au3Cu alloy. However, their network points are respectively (=51.55%, σ=0.64) and (=25%, σ=1) at 0 K. 3) The equilibrium path for a given composition (x) alloy is the standard path for studying subequilibrium  paths. These phenomena can not be expected in the QMAC- and CALPHAD-thermodynamics.
paths. These phenomena can not be expected in the QMAC- and CALPHAD-thermodynamics.
From Fig. 3(c), we have discovered surprising phenomena: All experimental middle jumping Tj-temperatures (denote by symbols “・” and “*” introduced from Ref. [35], which were erroneously considered as the so-called Tc-critical temperatures of equilibrium order-disorder transition of alloys, approach to equilibrium iso-order degree  curve.
curve.
Figure 3(d) shows that the network points of alloys with jumping-phenomena of order-disorder transition should be situated in the range from A-point to B-point, i.e., 0.755≤σ≤1 and 18.875%≤≤43.375%.
3.4 AG-concentration EHNP diagrams
From Fig. 4, the following main understandings may be obtained. 1) The AG-concentration EHNP diagrams, which are used to describe the AGA-structures of alloy phases, may be described by two modes: three-dimensional 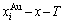 and
and  network phase diagrams (Figs. 4(a) and (b)) in the AGA- crystallography (see Appendix A.1), where the
network phase diagrams (Figs. 4(a) and (b)) in the AGA- crystallography (see Appendix A.1), where the 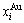 and
and  are the probabilities occupied at the lattice points; three-dimensional
are the probabilities occupied at the lattice points; three-dimensional 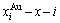 and
and  network phase diagrams (Figs. 4(c) and (d)) in the AGA-Gibbs energy level theory, where the and are the probabilities occupied at the and energy levels. 2) There exists an emergent phenomenon of some AG-concentrations in the ordered alloy phases, which are defined as that some AG-concentrations in the ordered state are larger than those in disorder state, such as
network phase diagrams (Figs. 4(c) and (d)) in the AGA-Gibbs energy level theory, where the and are the probabilities occupied at the and energy levels. 2) There exists an emergent phenomenon of some AG-concentrations in the ordered alloy phases, which are defined as that some AG-concentrations in the ordered state are larger than those in disorder state, such as 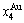 ,
, 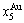 ,
, 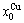 and
and 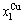 (Figs. 4(c), (d), (e) and (f)). 3) The equilibrium and paths for a given composition (x) alloy may be described by three-dimensional and equilibrium path charts (Figs. 4(g) and (h)) or two-dimensional
(Figs. 4(c), (d), (e) and (f)). 3) The equilibrium and paths for a given composition (x) alloy may be described by three-dimensional and equilibrium path charts (Figs. 4(g) and (h)) or two-dimensional 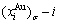 and
and  , as well as
, as well as  and
and  equilibrium path charts. The essential on disordering compound is that the
equilibrium path charts. The essential on disordering compound is that the 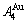 and
and  stem alloy genes are split into and
stem alloy genes are split into and 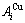 sequences in the disordered state. 4) It can be known that each kind of q-EHNP diagram includes not only the four type diagrams indicated above, but also other type diagrams. These AG-concentration equilibrium path charts will provide standard path charts for studying kinetic mechanism of experimental subequilibrium order-disorder transition path.
sequences in the disordered state. 4) It can be known that each kind of q-EHNP diagram includes not only the four type diagrams indicated above, but also other type diagrams. These AG-concentration equilibrium path charts will provide standard path charts for studying kinetic mechanism of experimental subequilibrium order-disorder transition path.
3.5 EHNP diagrams of other thermodynamic properties
According to and EHNP diagrams obtained from 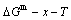 diagram, we have obtained q-x-T EHNP diagrams of other thermodynamic properties of Au3Cu-type sublattice system, which are shown in Appendix C. It should be emphasized that from each three-dimensional q-x-T EHNP diagram, we can obtain isocompositional isoproperty and isothermal path phase diagrams. The diagrams from Section 3.3 through present section are interconnected to form a big holographic network information database about structures, properties and their variations in composition and temperature of alloys. Therefore, the man’s knowledge of relationships of structures, properties and environments for alloys has been changed from single causality to systematic correlativity. Once one network point in any EHNP diagram above has been clicked, the information about composition, temperature, order degree, AGA-structure and a set of thermodynamic properties of the alloy as well as its equilibrium order-disorder transition EHNP charts may be readily obtained, which are very useful for materials engineers to design advanced alloys.
diagram, we have obtained q-x-T EHNP diagrams of other thermodynamic properties of Au3Cu-type sublattice system, which are shown in Appendix C. It should be emphasized that from each three-dimensional q-x-T EHNP diagram, we can obtain isocompositional isoproperty and isothermal path phase diagrams. The diagrams from Section 3.3 through present section are interconnected to form a big holographic network information database about structures, properties and their variations in composition and temperature of alloys. Therefore, the man’s knowledge of relationships of structures, properties and environments for alloys has been changed from single causality to systematic correlativity. Once one network point in any EHNP diagram above has been clicked, the information about composition, temperature, order degree, AGA-structure and a set of thermodynamic properties of the alloy as well as its equilibrium order-disorder transition EHNP charts may be readily obtained, which are very useful for materials engineers to design advanced alloys.
4 Discussion
4.1 Gibbs energies of stoichiometric compounds
It seems to be a fundamental concept prevailing in QMAC- and CALPHAD-thermodynamics that a stoichiometric compound should have the lowest Gibbs energy and highest critical temperature in its ordered sublattice system. From Fig. 5, it can be known that the lowest points on the  ,
, 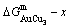 and
and 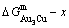 curves are respectively the L2-point, which is exactly equal to C2-point of the stoichiometric
curves are respectively the L2-point, which is exactly equal to C2-point of the stoichiometric  compound, the L3-point, which is slightly deviated from the C3-point of the stoichiometric
compound, the L3-point, which is slightly deviated from the C3-point of the stoichiometric  compound, and the L1-point, which is notably deviated from the C1-point of the stoichiometric
compound, and the L1-point, which is notably deviated from the C1-point of the stoichiometric  compound. These calculated results have never been obtained by researchers in the QMAC- and CALPHAD-communities. From their calculated phase diagrams, it can be known that they have consistently thought that stoichiometric Au3Cu compound should have the lowest Gibbs energies and highest critical temperatures in their ordered sublattice systems [14,25-27,36].
compound. These calculated results have never been obtained by researchers in the QMAC- and CALPHAD-communities. From their calculated phase diagrams, it can be known that they have consistently thought that stoichiometric Au3Cu compound should have the lowest Gibbs energies and highest critical temperatures in their ordered sublattice systems [14,25-27,36].
4.2 Some unexpected phenomena
In an extensive X-ray investigation of the Au-Cu system performed by LU and LIANG [37-39], some wondering phenomena were obtained, that may be explained as follows (see Fig. 5).
1) The composition range of the ordered Au3Cu- type alloys varies with annealing times. Under the experimental condition of six months of heat treatment, the composition range is ①: B-D, which does not include the stoichiometric Au3Cu compound. This wondering phenomenon has been predicated by our research results. From Fig. 5, it can be known that, the ①: B-D range is included in the theoretical limit range J1-J2 (18.875%-43.375% Cu) of long range order (LRO) Au3Cu-type alloys, which is determined by the first jumping order degree (0.755) of the jumping 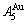 -alloy gene (see Fig. 3(d)); the lowest composition L1-point on the curve is notably deviated from the C1-point of the stoichiometric Au3Cu compound; it possesses higher Gibbs energy than the alloys in the ①: B-D range, and the reaction rate is very slow as well.
-alloy gene (see Fig. 3(d)); the lowest composition L1-point on the curve is notably deviated from the C1-point of the stoichiometric Au3Cu compound; it possesses higher Gibbs energy than the alloys in the ①: B-D range, and the reaction rate is very slow as well.
2) Under the experimental condition of one year of heat treatment, the composition range is ②: A-E, which includes the stoichiometric Au3Cu compound. Here, the lower composition B-point reaches the A-point, which is near to the composition J1-limit; the upper composition E-point reaches the cross O1-point of the and curves, which is far away from the upper composition J2-limit determined by the Au3Cu-type sublattice system, due to the stop of the AuCu-type sublattice system. We may also predicate that if increasing the time of heat treatment, the lower composition point would move more close to the lower composition J1-limit, but the upper composition point would not move, due to the existence of the AuCu-type sublattice system with lower curve.
3) According to analogous analysis, we have predicated that the theoretical limit composition range of the LRO-AuCu3-type alloys in the single AuCu3-type sublattice system should be from J3-point to J4-point [14], and its limit composition range in the Au-Cu system should be from the O2-point to J4-point, where the O2-point is the cross point of the and curves (see Fig. 5); as well as that the lowest L2-point on the curve at 0 K is near to the C3-point. These predicated results are in good agreement with the experiment phenomena (see Fig. 6).

Fig. 6 Limit composition ranges of long range ordered Au3Cu-type, AuCu-type and AuCu3-type alloys in Au-Cu system determined by their first jumping order degrees together with experimental jumping points, which are introduced from Ref. [25]
4) Figure 5 shows that the C1-C2 and C2-C3 composition ranges are respectively the theoretical equilibrium of two phase regions between the ordered Au3Cu-type and AuCu-type phases and between the ordered AuCu-type and AuCu3-type phases. However, the experimental measurements show that the two-phase coexistences disappear after one year’s treatment. Therefore, the experimental single Au3Cu-type alloys in the C1-O1 range, single AuCu-type alloys in the O1-O2 range and single AuCu3-type alloys in the O2-C3 range are really in a metastable state.
5) Finally, the experimental measurements show that there exist no two-phase regions between the ordered Au3Cu-type phase and the disordered phase and between the ordered AuCu3-type phase and the disordered phase at room temperature, that has been demonstrated by difference method of Gibbs energies between ordered and disordered phases (see Appendix B).
5 Conclusions
1) Based on the AG-holographic information database and essential definition of equilibrium order-disorder transition, the EHNP-diagrams of the Au3Cu-type sublattice have been established by the minimal mixed Gibbs energy path method. These diagrams exhibit unexpected characteristics of equilibrium transition of Au3Cu-type sublattice system, and may be used as a standard for studying experimental subequilibrium transition. Once one network point has been clicked, the information about the composition, temperature, AG-concentrations, holographic properties and EHNP-charts of the alloy may be readily obtained. These achievements will prove stimulating to materials engineers, and who may well find value in using it as a big information database for materials discovery, design, manufacture and application.
2) To establish AG-Gibbs energy partition function should obey six rules, which can make us to understand problematic essentials of the currently used alloy solution models in the QMAC-thermodynamics and CALPHAD-thermodynamics.
3) It has been proved that the AG-generalized vibration entropy energy is always larger than the AG-generalized vibration energy 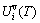 and that the AG-probabilities occupied at the AG-Gibbs energy levels in the degeneracy function for calculating configurational entropy should be degenerated by thoes of the constituent units occupied at the sublattice points, i.e., Bragg-William model.
and that the AG-probabilities occupied at the AG-Gibbs energy levels in the degeneracy function for calculating configurational entropy should be degenerated by thoes of the constituent units occupied at the sublattice points, i.e., Bragg-William model.
4) In the Au-Cu system, the experimental lower limit composition of the LRO-Au3Cu-type alloys is determined by the first jumping order degree, and its upper limit composition is determined by the cross point of the 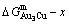 and curves at 0 K; the experimental lower limit composition of the LRO-AuCu3-type alloys is determined by the cross point of the and
and curves at 0 K; the experimental lower limit composition of the LRO-AuCu3-type alloys is determined by the cross point of the and  curves at 0 K, and its upper limit composition is determined by the first jumping order degree, and the experimental lower limit composition range of the LRO-AuCu-type alloys is determined by two cross points above. However, the heat treatment conditions and heating rates have great influence on jumping temperatures.
curves at 0 K, and its upper limit composition is determined by the first jumping order degree, and the experimental lower limit composition range of the LRO-AuCu-type alloys is determined by two cross points above. However, the heat treatment conditions and heating rates have great influence on jumping temperatures.
5) Today’s researchers in metallic materials science and physics do not understand that a real Gibbs energy function should be derived from the real Gibbs energy partition function constructed of the basic structure unit sequences and their Gibbs energy sequences. It is the fourth barrier to hinder the progress of the metal materials science, because some problems existed in these so-called Gibbs energy functions can not be easily overcome and the reductionism is still in the dominate position.
Appendixes
A Explanations of rules for establishing alloy gene Gibbs energy parathion function
A.1 Explanation of constituent units and structure units
In the traditional crystallography of alloy phases, the smallest representative of a crystal is a cell, which is described by taking symmetry elements to operate constituent atoms as structure units. Therefore, it may be named the constituent atom arranging (CAA) structure of alloy phase. Table A.1 shows the CAA-structure of the Au- and Cu-pure metals and alloy phases. In the Au- and Cu-pure metals, stoichiometric AuCu-, AuCu3- and Au3Cu-compounds, AuCu-type, AuCu3-type and Au3Cu-type ordered alloys, as well as disordered alloys, all lattice points are occupied by Au- and Cu-constituent atoms. We can not know their information about coordinative configurations, electronic structures, potential energies, vibration energies and volumes, as well as other properties. Therefore, the Au- and Cu-constituent atoms belong in degenerated atoms respectively by the and characteristic atoms and incomplete sequences with less information about only properties of Au-and Cu-pure metals and some unknown energetic parameters. The Au- and Cu-pure metals belong in degenerated crystals respectively by the  and
and  characteristic crystals. Therefore, the alloy may be defined as the CAA-solution.
characteristic crystals. Therefore, the alloy may be defined as the CAA-solution.
In the AGA-crystallography of the SMMS framework, the crystalline cell is described by taking symmetry elements operate alloy genes (i.e., characteristic atoms) as structure unit sequences. Table A.2 gives the AGA-structures of the Au- and Cu-pure metals, stoichiometric AuCu-, AuCu3- and Au3Cu-compounds, AuCu-type, AuCu3-type and Au3Cu-type ordered alloys, as well as disordered alloys. The lattice points are occupied by and characteristic atoms. We can know their information about coordinative configurations, electronic structures, potential energies, vibration energies and volumes, as well as other properties [1]. The AGA-cells and potential wave planes of the AuCu-, AuCu3- and Au3Cu-compounds are shown in Fig. A.1 [39]. Therefore, the alloy may be defined as the AGA-solution.
A.2 Generalized vibration free energy and generalized vibration entropy energy
It has been proved that the AG-generalized vibration free energy  always is negative, because the generalized vibration entropy energy is always larger than the generalized vibration energy
always is negative, because the generalized vibration entropy energy is always larger than the generalized vibration energy 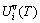 .
.
The contributions of AG-generalized vibration energies and AG-generalized vibration entropy energies to AG-Gibbs energies. The AG-Gibbs energy is a complex function with multi-energetic levels:
 (1)
(1)
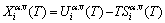 (2)
(2)
 (3)
(3)
 (4)
(4)
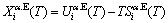 (5)
(5)
 (6)
(6)
Table A.1 CAA-crystal structures of alloy phases in Au-Cu system

Table A.2 AGA-crystal structures of alloy phases in Au-Cu system
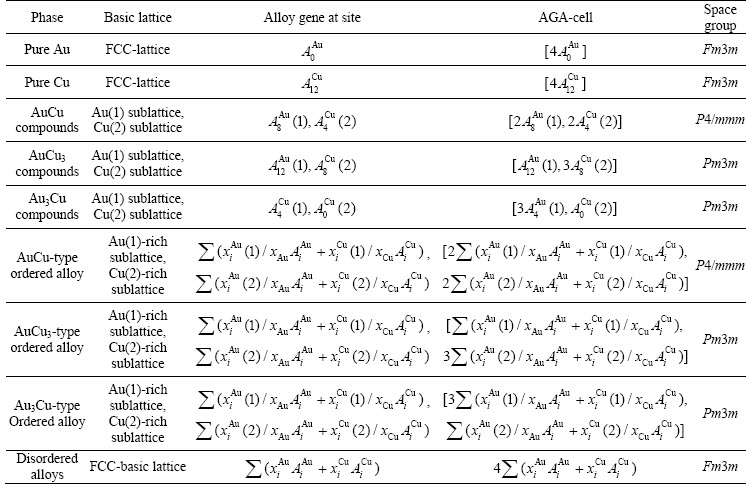
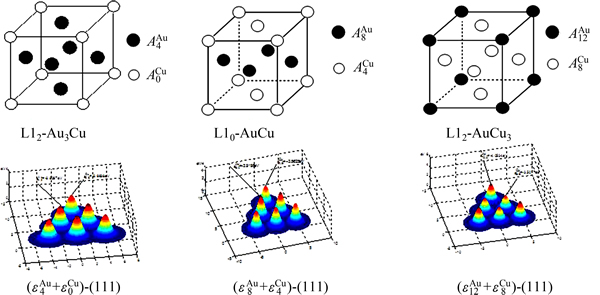
Fig. A.1 Characteristic atom arrangement cells and potential energy wave (111) planes of compounds L12-Au3Cu, L10-AuCu and L12-AuCu3
 (7)
(7)
 (8)
(8)
 (9)
(9)
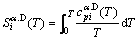 (10)
(10)
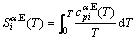 (11)
(11)
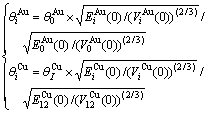 (12)
(12)
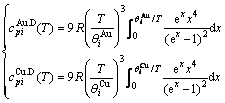 (13)
(13)
 (14)
(14)
 (15)
(15)
where α denotes Au or Cu; R is the gas constant; 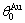 =165 K,
=165 K,  =175.21 K,
=175.21 K, 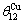 =343 K,
=343 K, 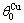 =343.07 K.
=343.07 K.
It has been demonstrated that the q-energetic property sequence in the Au-Cu system may be described by following equation:
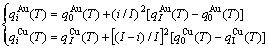 (16)
(16)
From Tables A.3 to A.6, therefore, it can be known that the so-called equilibrium order-disorder transition  path obtained by the AGA-potential free energy function of the alloy is higher than the
path obtained by the AGA-potential free energy function of the alloy is higher than the 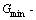 path obtained by the AGA-Gibbs free energy function, because the
path obtained by the AGA-Gibbs free energy function, because the  is always lower than the corresponding
is always lower than the corresponding  and the
and the  is always larger than the corresponding
is always larger than the corresponding 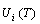 .
.
A.3 Alloy gene Gibbs energy levels and alloy gene pair Gibbs energy levels
Analogous with AG-Gibbs energy partition function, the AG-pair Gibbs energy partition function may be used to describe systematic correlativity of the AG-pair Gibbs energy levels, the AG-cluster probabilities occupied at the AG-pair Gibbs energy levels and degeneracy function of these probabilities as function of composition (x), temperature (T) and order degree (σ), and to derive the Gibbs energy function.
In the Au-Cu system, the complete AG-pair sequences contain 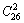 =325 kinds of AG-pairs, which are
=325 kinds of AG-pairs, which are 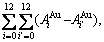
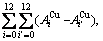
 and the complete AG-pair Gibbs energy sequences contain =325 kinds of AG-pairs Gibbs energy levels, which are
and the complete AG-pair Gibbs energy sequences contain =325 kinds of AG-pairs Gibbs energy levels, which are 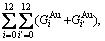

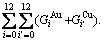 The AG-pair probabilities (i.e., concentrations) occupied at the AG-pair Gibbs energy levels are respectively
The AG-pair probabilities (i.e., concentrations) occupied at the AG-pair Gibbs energy levels are respectively 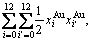

 Therefore, the AG-pair Gibbs energy partition function may be established and the characteristic Gibbs energy of an alloy should be
Therefore, the AG-pair Gibbs energy partition function may be established and the characteristic Gibbs energy of an alloy should be
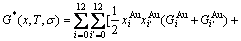
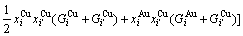 (17)
(17)
It may be proved that Eqs. (2) and (17) are equivalent. For the Au3Cu () compound with =75% Au and =25% Cu, its molar characteristic Gibbs energy calculated by Eq. (17) is
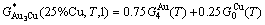
For the AuCu() compound with  = 50% Au and
= 50% Au and 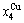 =50% Cu, its molar characteristic Gibbs energy calculated by Eq. (17) is
=50% Cu, its molar characteristic Gibbs energy calculated by Eq. (17) is

For the AuCu3  compound with
compound with  = 25% Au and
= 25% Au and  =75% Cu, its molar characteristic Gibbs energy calculated by Eq. (17) is
=75% Cu, its molar characteristic Gibbs energy calculated by Eq. (17) is
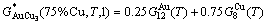
Analogous with AG-Gibbs energy partition function, the AG-cluster Gibbs energy partition function may be used to describe systematic correlativity of the AG- cluster Gibbs energy levels, the AG-cluster probabilities occupied at the AG-cluster Gibbs energy levels and degeneracy function of these probabilities as function of composition (x), temperature (T) and order degree (σ), and to derive the Gibbs energy function. However, for a given alloy, its characteristic Gibbs energies , configuration entropy 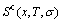 and Gibbs energies
and Gibbs energies 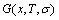 calculated respectively by AG-, AG-pair and by AG-cluster sequence as structure unit sequences should be identical, because , and functions are interrelated. For simplified calculation, we should adopt AG-sequence as structure unit sequence, rather than AG-pair sequence and AG-cluster sequences. It has been also demonstrated that the configurational entropy is independent on sizes of structure units, if they are complete sequences.
calculated respectively by AG-, AG-pair and by AG-cluster sequence as structure unit sequences should be identical, because , and functions are interrelated. For simplified calculation, we should adopt AG-sequence as structure unit sequence, rather than AG-pair sequence and AG-cluster sequences. It has been also demonstrated that the configurational entropy is independent on sizes of structure units, if they are complete sequences.
Table A.3  ,
, 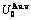 ,
, 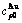 ,
,  , (
, (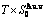 ),
),  ,
,  and
and 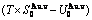 of
of 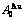 -alloy gene
-alloy gene
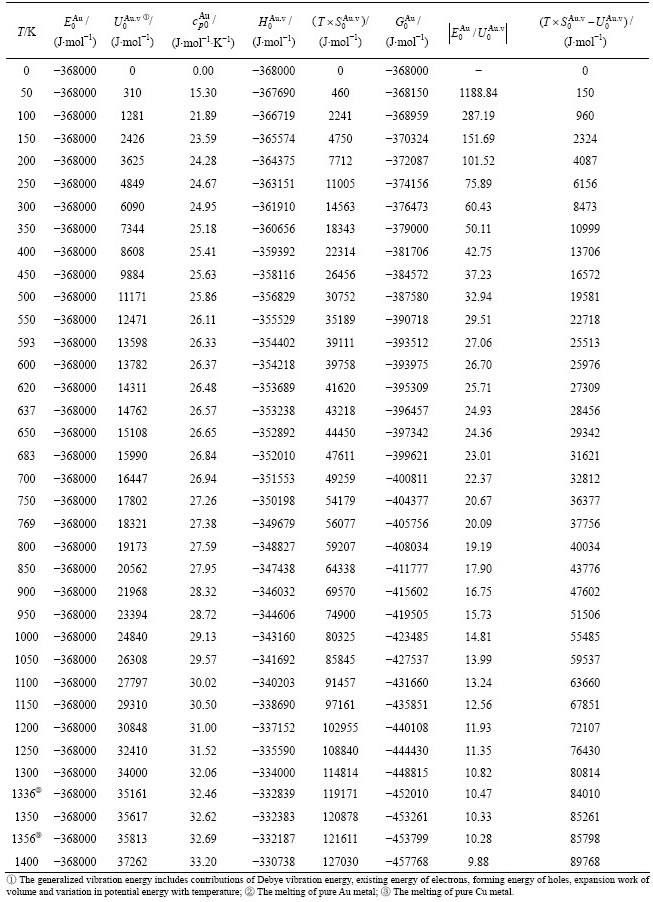
Table A.4 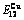 ,
,  ,
,  ,
,  , (
, (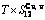 ),
),  ,
, 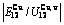 and
and 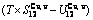 of
of 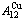 -alloy gene
-alloy gene
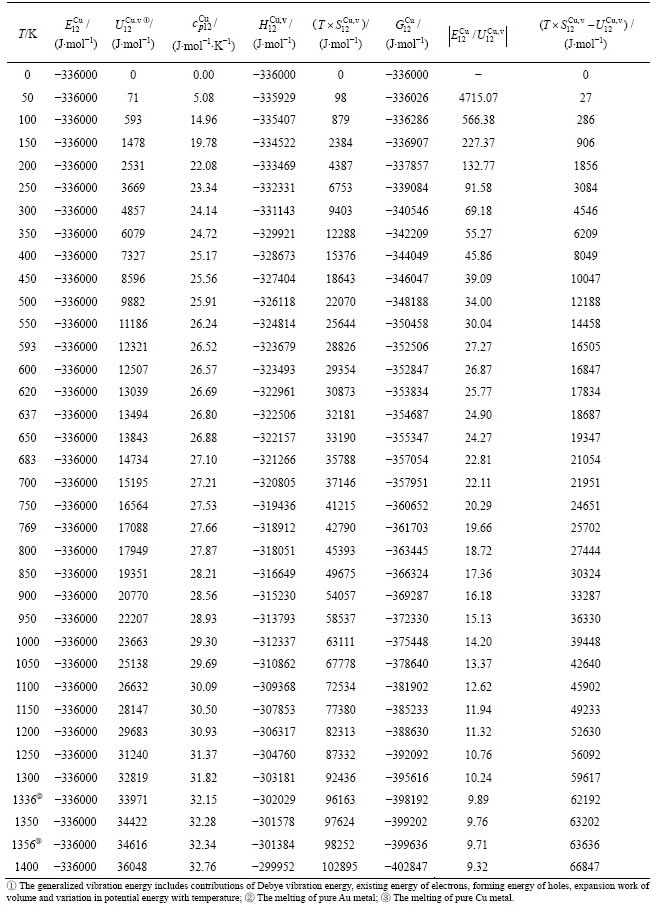
Table A.5  ,
,  ,
, 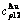 ,
,  , (
, (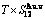 ),
),  ,
,  and
and  of
of  -alloy gene
-alloy gene
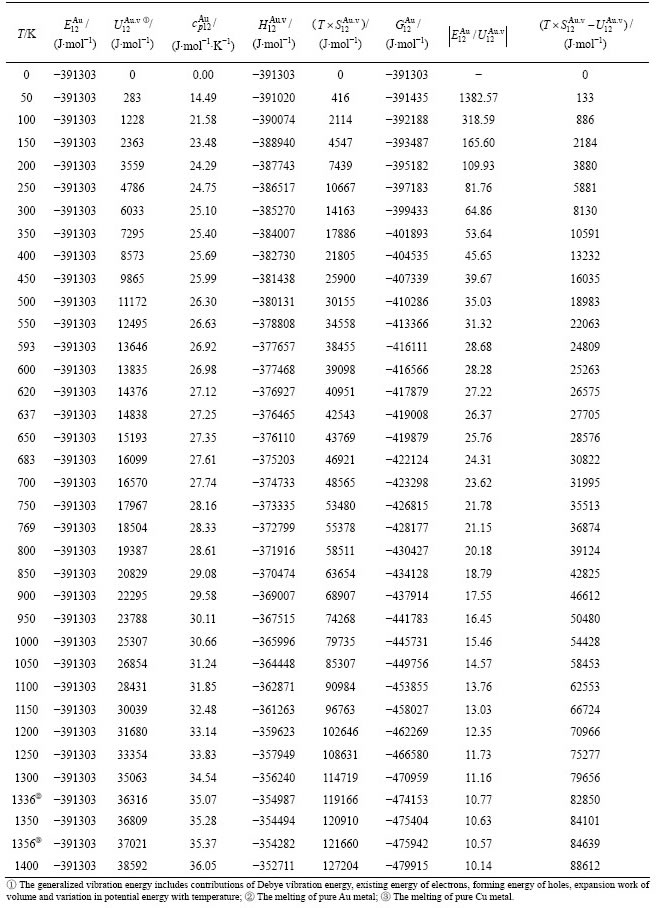
Table A.6  ,
,  ,
,  ,
,  , (
, ( ),
),  ,
,  and
and  of the
of the  -alloy gene
-alloy gene
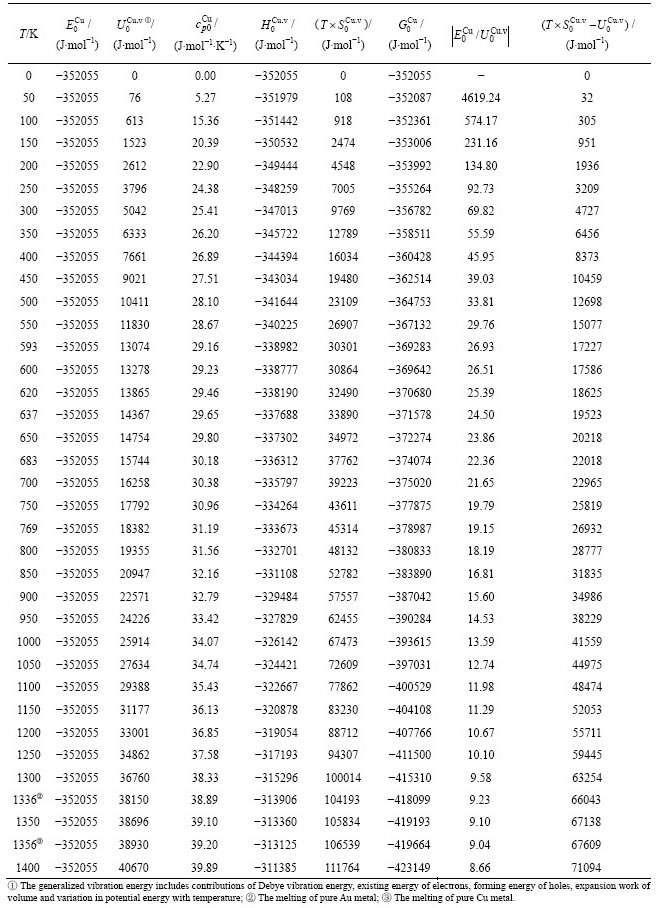
A.4 Configurational entropy and configurational entropy energy
In the SMMS-framework, the AG-Gibbs energy function derived from the AG-Gibbs energy partition function in the Au-Cu system is a complex function with multi-levels:
 (18)
(18)
The following understandings have been obtained:
1) Once the AG-holographic information database of the fcc lattice based Au-Cu system has been established, the AG- function can be used to described the ordered alloys in the Au3Cu-, AuCu- and AuCu3-type sublattice systems and disordered alloy, where there is no unknown parameter.
function can be used to described the ordered alloys in the Au3Cu-, AuCu- and AuCu3-type sublattice systems and disordered alloy, where there is no unknown parameter.
2) The AG-propabilities occupied at the AG-Gibbs energy levels of the equilibrium order- disorder transition for a given alloy are determined by the minimum mixed Gibbs energy path method [1].
occupied at the AG-Gibbs energy levels of the equilibrium order- disorder transition for a given alloy are determined by the minimum mixed Gibbs energy path method [1].
3) The AG-propabilitiesoccupied at the AG-Gibbs energy levels of the subequilibrium order- disorder transition for a given alloy are determined by the experimental mixed enthalpy 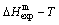 tracking path method [16].
tracking path method [16].
4) The 
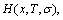

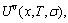
 and
and  functions are internected. It means that the configurational entropy function can not be independently treated.
functions are internected. It means that the configurational entropy function can not be independently treated.
5) According to the first rule, it has been known that the constituent atoms, constituent atom pairs and constituent atom clusters can not be used as structure unit sequences, because they are incomplete sequences and have no their Gibbs energy levels.
6) According to the second rule, it has been known that the established so-called Gibbs energy functions with a number of unknown parameters by the constituent atoms, constituent atom pairs and constituent atom clusters are too crude in theory, and that based on the miss understandings of experimental information, these parameters are adjusted until the Gibbs energy function is capable of representing the experimental information, which is also questionable in practice.
7) According to the third and fourth rules, it has been known that if using AG-pairs with AG-pairs Gibbs energy levels and AG-clusters with AG-cluster Gibbs energy levels as structure units sequences, the calculated total Gibbs energies, characteristic Gibbs energies, potential energies, generalized vibration energies, generalized vibration entropies and configurational entropies are still identical respectively with ones obtained by the AG-sequences. It means that the configurational entropy is independent on sizes of structure units.
In order to understand how to calculate the configurational entropy from the AG-propabilities occupied at the AG-Gibbs energy levels, we have calculated the EHNP-diagrams of the Au3Cu-, AuCu-, AuCu3-type sublattice systems by Eqs. (19)-(24), which are shown in Figs. A.2-A.5. In our opinion, the AG-propabilities occupied at the AG-Gibbs energy levels in the degeneracy g-function should be degenerated by the ones of the constituent atoms occupied at the lattice points, based on following reasons:
1) The generalized vibration entropy has been accounted in the  -characteristic Gibbs energy (see Eq. (18)). Therefore, the configurational entropy , arisen from the AG-propabilities occupied at the lattice points should be degenerated by ones of the constituent atoms occupied at the lattice points.
-characteristic Gibbs energy (see Eq. (18)). Therefore, the configurational entropy , arisen from the AG-propabilities occupied at the lattice points should be degenerated by ones of the constituent atoms occupied at the lattice points.
2) The configurational entropy of each ordered alloy calculated by a degeneracy function can change continually from the configurational entropy in the maximum order degree state to one in the perfectly disordered state.
3) For binary alloy systems, the should lie in the range:
0≤≤5.763 (J・mol-1・K-1)
where the upper limit is the value for a perfectly disordered equiatomic alloy.
The results show that the AGA-degeneracy function (1) and entropy function (1) may be used. The calculated details may be seen in Refs. [1,40,41].
The AGA degeneracy function (1) and entropy function (1):

Fig. A.2 Configuration entropy EHNP diagrams of Au3Cu-type sublattice system systems calculated by 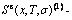 function
function
 (19)
(19)

 (20)
(20)
The AGA degeneracy function (2) and entropy function (2):

 (21)
(21)

 (22)
(22)
The AGA degeneracy function (3) and entropy function (3):

 (23)
(23)

 (24)
(24)
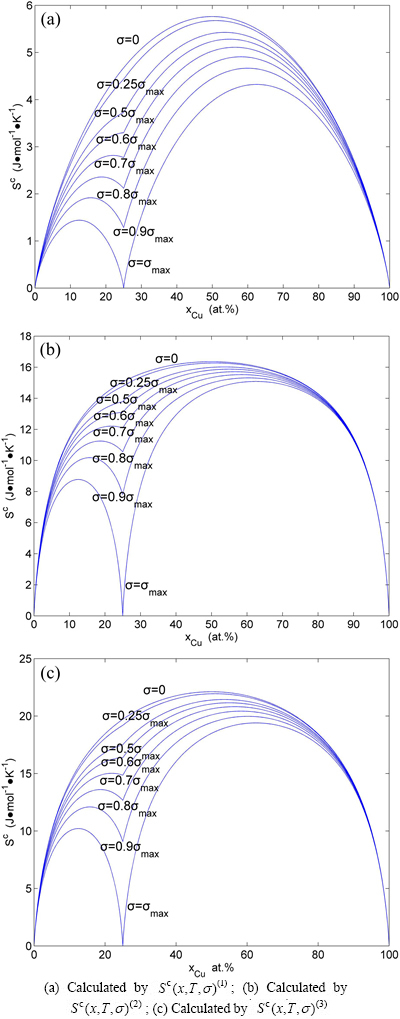
Fig. A.3 Congfiguration entropies of Au3Cu-type sublattice system
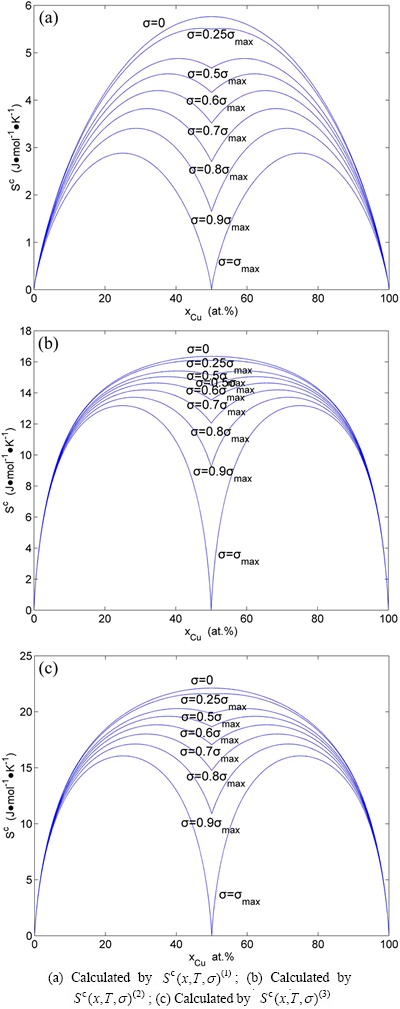
Fig. A.4 Congfiguration entropies of AuCu-type sublattice system
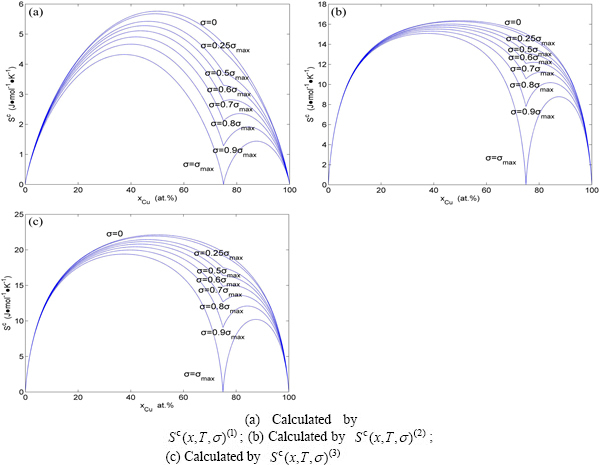
Fig. A.5 Congfiguration entropies of AuCu3- type sublattice system
B EHNP-charts on disordering stoichiometric Au3Cu compound
According to and paths on disordering stoichiometric Au3Cu compound obtained by the minimal mixed Gibbs energy method, the EHNP charts are calculated and shown in Figs. B.1 to B.13. By the same method, the systematic correlativity data of the , , , and on equilibrium order-disorder transition paths as function of composition and temperature for alloys of the Au3Cu-type sublattice system are calculated, using calculated steps  =0.5%,
=0.5%,  =1 K,
=1 K,  = 0.0001.
= 0.0001.
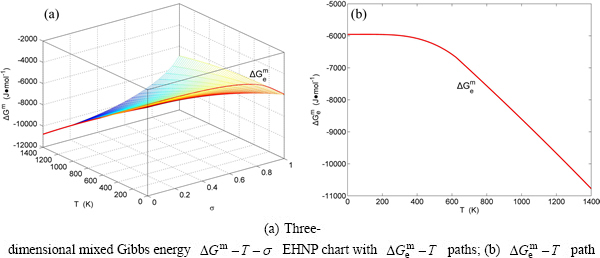
Fig. B.1 EHNP charts with EHNP curve of first order thermodynamic properties on disordering 
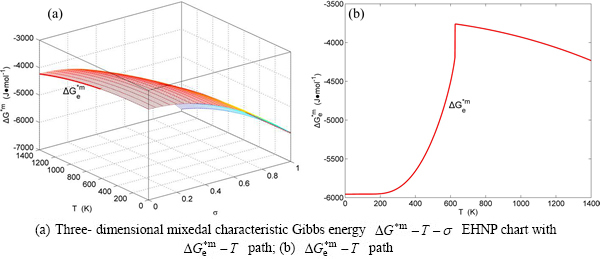
Fig. B.2 EHNP charts with EHNP curve of first order thermodynamic properties on disordering
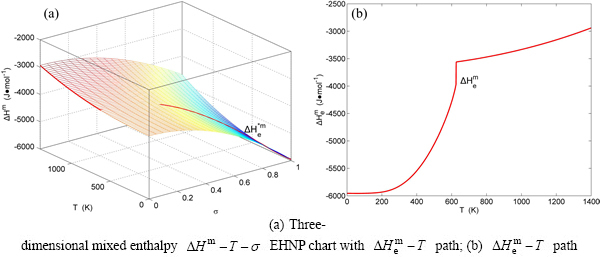
Fig. B.3 EHNP charts with EHNP curve of first order thermodynamic properties on disordering
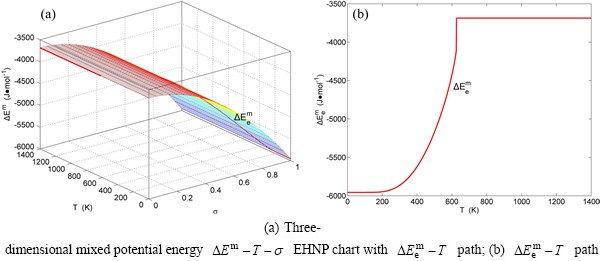
Fig. B.4 EHNP charts with EHNP curve of first order thermodynamic properties on disordering

Fig. B.5 EHNP charts with EHNP curve of first order thermodynamic properties on disordering

Fig. B.6 EHNP charts with EHNP curve of first order thermodynamic properties on disordering
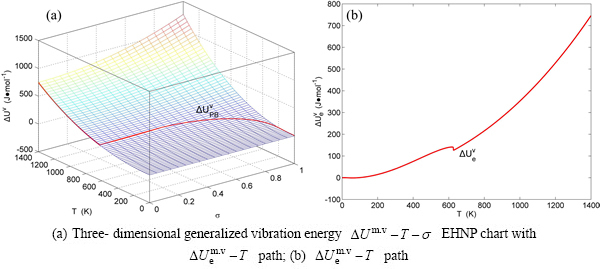
Fig. B.7 EHNP charts with EHNP curve of first order thermodynamic properties on disordering
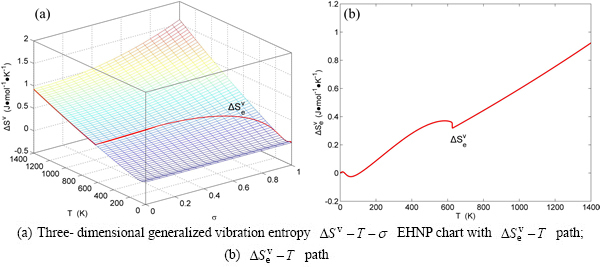
Fig. B.8 EHNP charts with EHNP curve of first order thermodynamic properties on disordering
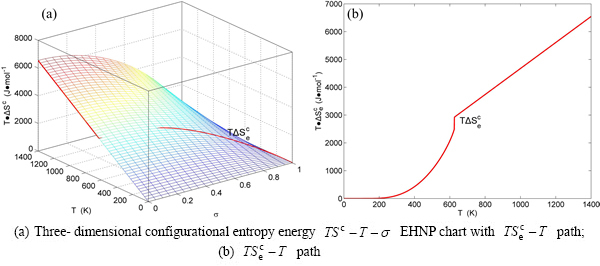
Fig. B.9 EHNP charts with EHNP curve of first order thermodynamic properties on disordering

Fig. B.10 EHNP charts with EHNP curve of first order thermodynamic properties on disordering

Fig. B.11 EHNP charts with EHNP curve of first order thermodynamic properties on disordering

Fig. B.12 Second order thermodynamic properties (heat capacity and thermal expansion coefficient) on disordering
C Difference method of Gibbs energies between ordered and disordered phases
The -phase boundary curve of Au3Cu- type sublattice system has been obtained by the difference method of Gibbs energies between ordered Au3Cu-type phase and disordered phase (see Fig. C.1). It has been proved that there is no two-phase region of the ordered and disordered phases, because ordered and disordered alloys belong in the same fcc-based lattice Au-Cu system.
D Other thermodynamic properties EHNP diagrams
According to and 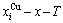 EHNP diagrams obtained from diagram, we have obtained other
EHNP diagrams obtained from diagram, we have obtained other 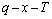 EHNP diagrams of AuCu3- type sublattice system shown in Figs. D.1 and D.2. It should be emphasized that from each three-dimensional EHNP diagram, we can obtain isocompositional
EHNP diagrams of AuCu3- type sublattice system shown in Figs. D.1 and D.2. It should be emphasized that from each three-dimensional EHNP diagram, we can obtain isocompositional , isoproperty
, isoproperty and isothermal
and isothermal  path phase diagrams. These diagrams are interconnected to form a big database about structure, properties and their variations with temperature of alloy systems. Therefore, the man’s knowledge of relationships of structure, properties and environments for alloy systems has been changed from single causality to systematic correlativity.
path phase diagrams. These diagrams are interconnected to form a big database about structure, properties and their variations with temperature of alloy systems. Therefore, the man’s knowledge of relationships of structure, properties and environments for alloy systems has been changed from single causality to systematic correlativity.

Fig. B.13 Activities on disordering 
Fig. C.1 Difference method for calculating phase boundary curve of Au3Cu-type sublattice system
Fig. D.1 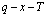 EHNP diagrams of Au3Cu-type sublattice system
EHNP diagrams of Au3Cu-type sublattice system
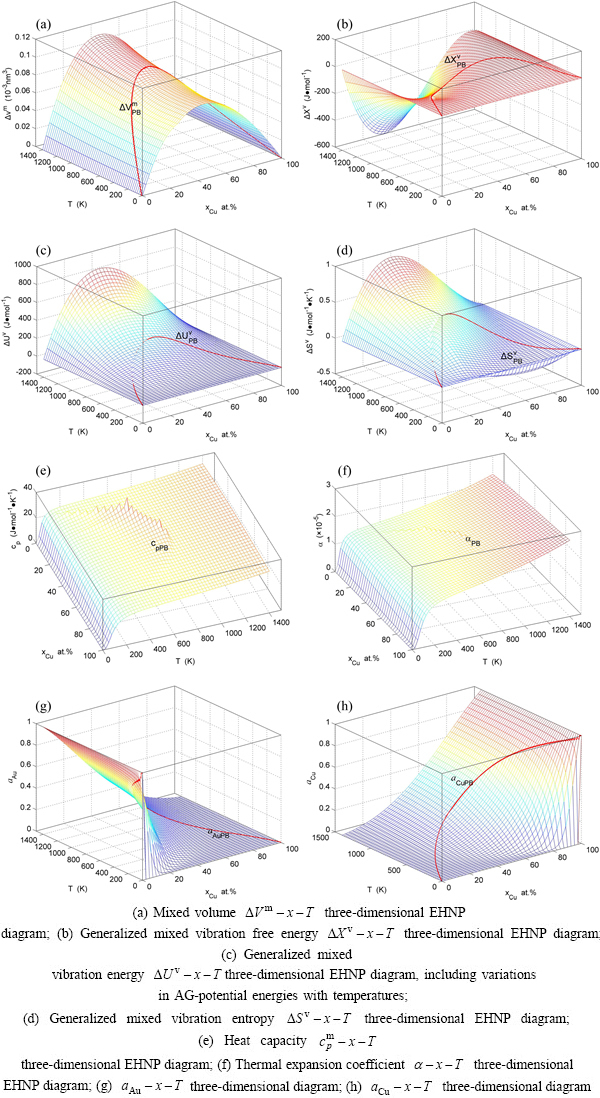
Fig. D.2 EHNP diagrams of Au3Cu-type sublattice system
References
[1] XIE You-qing, LI Xiao-bo, LIU Xin-bi, NIE Yao-zhuang, PENG Hong-jian. Alloy gene Gibbs energy partition function and equilibrium holographic network phase diagrams of AuCu-type sublattice system [J]. International Journal of Communications, Network and System Sciences, 2013, 12(6): 415-442.
[2] XIE You-qing. Systematic science of metallic materials [M]. Changsha: Central South University of Technology Press, 1998. (in Chinese)
[3] XIE You-qing. Systematic science of alloys [M]. Changsha: Central South University Press, 2012. (in Chinese)
[4] XIE You-qing. Atomic energies and Gibbs energy functions of Ag-Cu alloys [J]. Science in China, Series E: Technological Science, 1998, 41(2): 146-156.
[5] XIE You-qing, ZHANG Xiao-dong. Atomic volumes and volume functions of Ag-Cu alloys [J]. Science in China, Series E: Technological Science, 1998, 41(2): 157-168.
[6] XIE You-qing. A new potential function with many-atom interactions in solid [J]. Science in China, Series A, 1993, 36(1): 90-99.
[7] XIE You-qing. Electronic structure and properties of pure iron [J]. Acta Metallargics et Materialia, 1994, 42(11): 3705-3715.
[8] XIE You-qing, LIU Xin-bi, PENG Hong-jian, NIE Yao-zhuang, LI Xiao-bo, LI Yan-feng. Characteristic atom arranging crystallography of alloy phases for Au-Cu system [J]. Science China: Technological Sciences, 2011, 54(6): 1560-1567.
[9] XIE You-qing, PENG Kun, LIU Xin-bi. Influences of xTi/xAl atomic states, lattice constants and potential-energy planes of ordered fcc TiAl type alloys [J]. Physica B, 2004, 344(1-4): 5-20.
[10] XIE You-qing, LIU Xin-bi, PENG Kun. Atomic states, potential energies, volumes, stability and brittleness of ordered fcc TiAl3 type alloys [J]. Physica B, 2004, 353(1-2): 15-33.
[11] XIE You-qing, PENG Hong-jian, LIU Xin-bi, PENG Kun. Atomic states, potential energies, volumes, stability and brittleness of ordered fcc Ti3Al type alloys [J]. Physica B, 2005, 362(1-4): 1-17.
[12] XIE You-qing, TAO Hui-jin, PENG Hong-jian, LIU Xin-bi, PENG Kun. Atomic states, potential energies, volumes, stability and brittleness of ordered fcc TiAl2 type alloys [J]. Physica B, 2005, 366(1-4): 17-37.
[13] XIE You-qing, PENG Hong-jian, LIU Xin-bi, LI Xiao-bo, NIE Yao-zhuang. New atom movement mechanism for tracking path on disordering AuCuI( ) compound [J]. Transactions of Nonferrous Metals Society of China, 2014, 24(10): 3321-3256.
) compound [J]. Transactions of Nonferrous Metals Society of China, 2014, 24(10): 3321-3256.
[14] XIE You-qing, LI Xiao-bo, LIU Xin-bi, NIE Yao-zhuang, PENG Hong-jian. Alloy gene Gibbs energy partition function and equilibrium holographic network phase diagrams of AuCu3-type sublattice system [J]. Transactions of Nonferrous Metals Society of China, 2014, 24(11): 3585-3610.
[15]  B, PFEILER W. The retro-effect in stoichiometric CuAu: A resistometric study [J]. Intermetallics, 1997, 5(7): 501-505.
B, PFEILER W. The retro-effect in stoichiometric CuAu: A resistometric study [J]. Intermetallics, 1997, 5(7): 501-505.
[16] PFEILER W, B. Atomic ordering in alloys: Stable states and kinetics [J]. Materials Science and Engineering A, 2002, 324(1-2): 34-42.
[17] B, CHALUPA B. Checking the model for dynamic disordering in CuAu [J]. Materials Science and Engineering A, 2002, 324(1-2): 58-61.
[18] FOWLER R H, GUGGENHEIM E A. Statistical thermodynamics [M]. Cambridge: Cambridge University Press, 1937.
[19] MAYER J E, MAYER M G. Statistical mechanics [M]. New York: Wiley, 1940.
[20] GUGGENHEIM E A. Mixtures [M]. Oxford: Oxford University Press, 1952.
[21] PRIGOGINE I. Molecular theory of solutions [M]. Amsterdam: North Holland, 1957.
[22] HILL T L. Introduction to statistical thermodynamics [M]. Reading, MA: Addison-Wesley, 1960.
[23] XIE You-qing, ZHANG Xiao-dong. Electronic structures of Ag-Cu alloys [J]. Science in China, Series E: Technological Science, 1998, 41(3): 225-236.
[24] KIKUCHI R, de FONTAINE D, MURAKAMI M, NAKAMURA T. Ternary phase diagram calculations―II Examples of clustering and ordering systems [J]. Acta Metallurgica, 1997, 25(2): 207-219.
[25] SUNDMAN B, FRIES S G, OATES W A. A thermodynamic assessment of the Au-Cu system [J]. CALPHAD, 1998, 22(3): 335-354.
[26] SUNDMAN B, FRIES S G, OATES W A. A calphad assessment of the Au-Cu system using the cluster variatiation method [J]. Z Metallkd, 1999, 90(5): 267-273.
[27] CAO W, CHANG Y A, ZHU J, CHEN S, OATES W A. Thermodynamic modeling of the Cu-Ag-Au system using the cluster/site approximation[J]. Intermetallics, 2007, 15(8): 1438-1446.
[28]  V, WOLVERTON C, ZUNGER A. Cu-Au, Ag-Au, Cu-Ag, and Ni-Au Intermetallics: First-principles study of temperature-composition phase diagrams and structures [J]. Physical Review B, 1998, 57(12): 6427-6443.
V, WOLVERTON C, ZUNGER A. Cu-Au, Ag-Au, Cu-Ag, and Ni-Au Intermetallics: First-principles study of temperature-composition phase diagrams and structures [J]. Physical Review B, 1998, 57(12): 6427-6443.
[29] ASTA M, de FONTAINE D. First-principles study of phase stability of Ti-Al intermetallic compounds [J]. J Mater Res, 1993, 8(10): 2554-2568.
[30] DINSDALE A T. SGTE data for pure elements [J]. CALPHAD, 199l, 15(4): 317-425.
[31] KAUFMAN L,  J. First and second generation-birth of the materials genome [J]. Scripta Materialia, 2014, 70(1): 3-6.
J. First and second generation-birth of the materials genome [J]. Scripta Materialia, 2014, 70(1): 3-6.
[32] BRAGG W L, WILLIAMS E J. The effect of thermal agitation on atomic arrangement in alloys [J]. Proceedings the royal of society A, 1934, 145(2): 699-730.
[33] BRAGG W L, WILLIAMS E J. The effect of thermal agitation on atomic arrangement in alloys II [J]. Proceedings the Royal of Society A, 1935, 51(4): 540-566.
[34] XIE You-qing, MA Liu-ying, ZHANG Xiao-dong, ZHOU Ping, ZHAO Li-ying. Microstructure and properties of Cu-Ni alloy [J]. Science in China, Series A, 1993, 36(5): 612-623.
[35] OATES W A, ZHANG F, CHEN S L, CHANG Y A. Improved cluster-site approximation for the entropy of mixing in multicomponent solid solutions [J]. Physical Review B, 1999, 59(17): 11221-11225.
[36] WEI S H, MBAYE A A, FERRIRA L G, ZUNGER A. First-principles calculations of the phase diagrams of noble metals: Cu-Au, Cu-Ag and Ag-Au [J]. Phys Rev B, 1987, 36(20): 4163-4185.
[37] LU H S, LIANG C K. The superlattice formation and lattice spacing changes in copper-gold alloys [J]. Acta Physica Sinica, 1966, 22(6): 659-697. (in Chinese)
[38] LU H S, LIANG C K. The existence of the superlattice CuAu3 in the Cu-Au system [J]. Science Bulletin, 1966, 17(9): 395-396.
[39] LU H S, LIANG C K. Experimental evidence of order-disorder transitions as of the second degree [J]. Science Bulletin, 1966, 17(10): 441-443.
[40] XIE You-qing, LI Yan-fen, LIU Xin-bi, LI Xiao-bo, PENG Hong-jian, NIE Yao-zhuang. Characteristic atom occupation patterns of Au3Cu, AuCu3, AuCuI and AuCuII based on experimental data of disordered alloys [J]. Transactions of Nonferrous Metals Society of China, 2011, 21(5): 1092-1104.
[41] XIE You-qing, NIE Yao-zhuang, LI Xiao-bo, LIU Xin-bi, PENG Hong-jian, LI Yan-fen. Characteristic atom potential energy partition function of Au3Cu type ordered alloys in FP-SSA framework [J]. The Chinese Journal of Nonferrous Metals, 2011, 21(10): 2489-2501. (in Chinese).
谢佑卿1,2,3, 聂耀庄4,李小波5, 彭红建6, 刘心笔 1,2,3
1. 中南大学 材料科学与工程学院,长沙 410083;
2. 中南大学 粉末冶金研究院,长沙 410083;
3. 中南大学 粉末冶金国家重点实验室,长沙 410083;
4. 中南大学 物理与电子学院,长沙 410083;
5. 湘潭大学 材料科学与工程学院,湘潭 411105;
6. 中南大学 化学化工学院,长沙 410083
摘 要:以Au3Cu-亚格子系统为例,介绍了3项发现:第一,至今阻碍金属材料科学进步的第四大障碍是研究者们尚未认识到一个真正的合金相Gibbs能函数应由合金基因序列和它们自己的Gibbs能级序列构建的Gibbs能配分函数导出。第二,建立合金基因Gibbs能配分函数的六条规则,特别证明了计算合金组态熵的简并因子中结构单元占居Gibbs能级的概率应按照组元占居格点的概率方式简并;第三,以前研究者从未预料到的主要特征:具有一条没有有序相和无序相共存区的单相相界线;相界线顶点成分和温度远偏离Au3Cu化合物临界点的成分和温度;在0 K时,Gibbs能随成分变化曲线上的最低点成分远偏离Au3Cu化合物的成分;Au3Cu-型长程有序合金的理论极限成分范围由第一跳变有序度决定。
关键词:Au3Cu化合物;Au3Cu-型亚格子系统;合金基因Gibbs能配分函数;平衡全息网络相图;系统金属材料科学
(Edited by Yun-bin HE)
Foundation item: Project (51071181) supported by the National Natural Science Foundation of China; Project (2013FJ4043) supported by the Natural Science Foundation of Hunan Province, China
Corresponding author: You-qing XIE; Tel: +86-731-88879287; E-mail: xieyouq8088@163.com
DOI: 10.1016/S1003-6326(15)63598-1

