
Preparation, characterization and photo-catalytic behavior of WO3-TiO2 catalysts with oxygen vacancies
TONG Hai-xia(童海霞)1, CHEN Qi-yuan(陈启元)2, YIN Zhou-lan(尹周澜)2, HU Hui-ping(胡慧萍)2,
WU Dao-xin(吴道新)1, YANG Ya-hui(杨亚辉)2
1. Hunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation,
Chemical and Biologic Engineering Institute, Changsha Science and Technology University, Changsha 410076, China;
2. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
Received 10 April 2009; accepted 12 October 2009
Abstract: TiO2 photocatalysts compounded with WO3 were prepared via a modified hydrolysis process and the 2% WO3-TiO2 catalysts with different oxygen vacancies were obtained by calcination at 873 K in H2 atmosphere. The catalysts were identified using X-ray diffractometry (XRD), specific surface measurement (BET), electron paramagnetic resonance spectroscopy (EPR), UV-Vis diffusion refraction spectroscopy (DRS), and X-ray photoelectron spectroscopy (XPS). The photocatalytic activity of 2% WO3-TiO2 with different oxygen vacancies was investigated employing splitting of water for O2 evolution. The results indicate that appropriate oxygen vacancies can obviously improve the photocatalytic activity of 2% WO3-TiO2 catalysts, and using Fe3+ as an electron acceptor under UV irradiation in 12 h, the maximum rate for O2 evolution is 667 ?mol/(L・h).
Key words: TiO2; oxygen vacancies; photocatalyst; calcination
1 Introduction
Pure titania has low photo-activity and slow photolysis speed for photo-catalysis of water, and it is very difficult to be used practically[1]. So, it is necessary to improve its photo-activity and enhance the photolysis speed for water splitting. Generally, the photo-catalytic activity of semiconductors can be improved through doping, loading and compositing the semiconductor.
Most of these doped semiconductors were used to photo-decompose organic pollutants[2-4], and some of them were used to split water with low photo-catalytic efficiency[1] because the generated electron-hole pairs on the photocatalyst surface were easy to recombine. ZHANG et al[5] reported that WO3 thin films sputtered on TiO2 can improve the speed of the photo-catalytic degradation of methylene blue. However, there is no further report about its application in water splitting. In our previous work[6], WO3 compounding can improve the photo-catalytic activity of TiO2 and the optimum concentration of compounded WO3 is 2%. GAO et al[7] presented that calcination can change the band gap of WO3 using H2 as the reducing atmosphere. HARRIS and SCHUMACHER[8], and HELLER et al[9] found that calcination using H2 as the reducing atmosphere can prolong the life of the generated electron-hole pairs on the photocatalyst surface. CHEN et al[10], HOWE and GRATZEL[11], QIN et al[12], REKOSKE and BARTEAU[13] and LIU et al[14] reported that the oxygen vacancies(OV) and the Ti3+ generated after calcination using H2 as the reducing atmosphere are major factors for the improvement of the photo-catalytic activity of TiO2. However, most of the researchers only calcined the anatase TiO2 or WO3 in H2 atmosphere, and there is less further report about rutile TiO2 and compounding conductors calcined in H2 atmosphere. So, in the work, compounding conductor (2% WO3-TiO2) was calcined at 873 K in the H2 atmosphere to evaluate the effects of oxygen vacancies on the photo-catalytic activity. The reaction of photo-catalytic splitting water is carried out via two semi-reactions, of which one is the photo-reduction with H2 evolution, and the other is the photo-oxidation with O2 evolution with the latter being more difficult to achieve. Because of the low separation efficiency of photo-electrons and holes, the evolution rate of oxygen is low and it is difficult to match the rate of hydrogen evolution in the “Double-Bed” splitting water system. In the present work, in order to improve the photocatalytic activity of rutile TiO2, the catalyst, 2% WO3-TiO2, with oxygen vacancies is employed for the photocatalytic oxidation of water with O2 evolution.
2 Experimental
2.1 Synthesis of 2% WO3-TiO2 powder with oxygen vacancies
The compounded 2% WO3-TiO2 was synthesized according to our previous work[6]. The powder of 2% WO3-TiO2 was loaded in a navicular quartz vessel to be calcined at 873 K for 2, 3, 5, 7 and 9 h, respectively, in H2 atmosphere and the series of catalysts were titled as k1, k2, k3, k4 and k5 in turn. Especially, the catalyst 2% WO3-TiO2 which was calcined in air was titled as k0.
2.2 Characterization of catalysts
X-ray diffraction analysis (XRD) was used to check the coexistence of different crystal phases of the catalyst by a HATCHI D/max2250 powder X-ray diffractometer. The diffraction profiles were recorded with Cu Ka1 radiation (0.154 056 nm) over a 2θ range of 10?-90? operating at 40 kV and 300 mA. A plumbaginous counter with monochromator was used.
The pore size of 2% WO3-TiO2 with oxygen vacancies was obtained with a Quantachrome surface area and pore size analyzer (Quantachrome Corporation, Model NOVA-1000) using N2 as the exposed gas. Typically, 0.5-1.0 g of the catalysts was used for the measurement. The catalysts were outgassed at 300 ℃ before the nitrogen physical adsorption at 70 K.
Diffusing reflectance UV-Vis spectra (UV-DRS) measurements were carried out on a Purkinje TU-1901 UV/Vis spectrophotometer (Beijing, China) equipped with a diffusion reflectance accessory with an IS19-1 integrating sphere, and BaSO4 powder was used as reference.
The X-ray photoelectron spectra were examined to identify the surface state of the samples by a Kratos XSAM800 spectrometer with Mg Kα as a radiation source. The charging effect of XPS spectra was calibrated with a carbon peak at 284.7 eV as a standard.
The in situ electron paramagnetic resonance (EPR) measurement was performed using an Endor spectrometer (JEOL JES-F1XG) at the liquid nitrogen temperature of 77 K. A microwave with the frequency of 9.44 GHz was used and its power was set at 1 mW. The g factor was obtained by taking the signal of manganese as an internal standard. A specially designed cell in the in situ EPR measurement under H2 environment was used. After being thermally treated, the TiO2 samples were cooled to room temperature gradually, and then transferred together with the reactor into the resonance cavity of the EPR spectrometer still under the H2 environment.
The results were examined by HATCHI SP-2305 gas chromatography which was equipped with a thermal conductivity detector. The argon was used as the carrier gas and the fixed phase was molecule sieve (0.5 nm).
2.3 Photo-activity tests
Photo-oxidation experiments were conducted in a cylindrical photo reactor surrounded with a circulating water jacket as described in our previous work[6]. A light source (250 W high-pressure Hg lamp or 250 W Xe, Changsha Kexing, China) was covered with a glass jacket made of quartz (cutting off <200 nm). The reactor temperature was kept constant at 293 K using cooling water. A mixture of catalyst (2 g), distilled water (640 mL) and a required amount of Fe2(SO4)3 as electron acceptor were put into the reactor and then argon gas was introduced into the system to degas completely. The catalyst powder was suspended by using a magnetic stirrer. The product gas oppressed the liquid in the reactor into the graduated cylinder through the outlet, so the volume of product gas could be read through the graduated cylinder indirectly. The evolution of O2 was detected by gas chromatography.
3 Results and discussion
3.1 Characterization of catalyst
Fig.1 shows the XRD patterns for 2% WO3-TiO2 catalysts with oxygen vacancies. From the patterns, the TiO2 was rutile crystal, and it did not show the change of the structure of the TiO2 after being thermally treated in reducing atmosphere. From its peak shape, it could be seen that the crystal structure was already quite complete, and the characteristic peaks of rutile TiO2 were located at 2θ=27.56?, 36.21?, 41.38?, 54.44?, 56.74?, 69.09? corresponding to the (110), (101), (111), (211), (220) and (311) crystal faces, respectively, which were consistent with the characteristic peaks of TiO2 rutile completely[15]. At the same time, some distinct peaks for WO2 could be observed after being thermally treated for 2-7 h (k1, k2, k3, k4), and peaks for WO3 disappeared after being thermally treated for 9 h (k5).

Fig.1 XRD patterns of 2% WO3-TiO2 catalysts with oxygen vacancies
Fig.2 shows the EPR patterns for the 2% WO3-TiO2 catalysts with oxygen vacancies. Considering the fact that there are unpaired electrons in the structure of either Ti3+ or OV, the TiO2 samples treated with different calcination time were measured by the in situ EPR to determine their presence and alteration during the H2 reduction. The analytical results as shown in Fig.2 indicated that the peak with a g factor of 1.94 could be assigned to Ti3+, since it was very close to 1.946[16] and 1.989[11]. The peak with a g factor of 2.00 might be assigned to OV[15]. The very weak signals of Ti3+ and OV were presented for the sample k1(thermally treated for 2 h), and strong signals of Ti3+ and OV were presented for the sample k5 (thermally treated for 9 h).
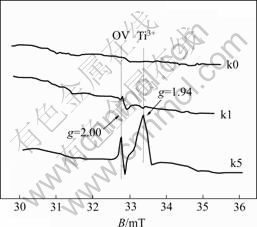
Fig.2 EPR patterns of 2% WO3-TiO2 catalysts with oxygen vacancies
Fg.3 shows the diffusion reflectance UV-Vis spectra of the catalysts k0, k1, k3, k4 and k5, and the pattern of k2 is deleted for its very little difference with k1. All patterns show obvious absorbing band-edge, which means that the samples have semiconductor characteristic. The absorbing band-edge near 413 nm means that the samples are rutile TiO2[17], which has a good agreement with the XRD results. The UV-Vis spectra of the catalysts show that the samples have the same absorbing ability for UV-light and the different absorbing ability for visible light. Just as XU[18] reported, when the metal oxide was thermally treated in reducing atmosphere, the following reactions occurred frequently:
OOx ――1/2O2 (g) + V o・+ e (1)
V o・――V o・・+ e (2)
where OOx is the initial status of the oxygen element in TiO2; Vo・, Vo・・ are the oxygen vacancies in the metal oxide. The oxygen vacancies in the metal oxide are the centers of the positive charges, which bound electrons easily. The electrons around OV are easily excited to the conduction band and thus they took the role of the donors. The band energy of the oxygen vacancies, ED, is near the base of the conduction band EC[19]. Longer time calcination makes more OV for the catalysts WO3-TiO2, which can band more electrons. So, in the range of 420-700 nm, the optical absorption properties of the catalysts become more strong with increasing the amount of oxygen vacancies (OV). Thus, longer time calcination means better absorption for visible light.
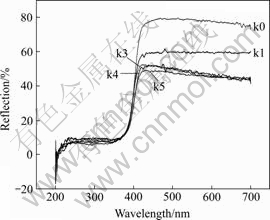
Fig.3 Diffusion reflectance UV-Vis spectra of 2% WO3-TiO2 catalysts with oxygen vacancies
The particle size distribution of the compounded 2% WO3-TiO2 with oxygen vacancies is shown in Fig.4. It can be seen that the particle size distribution of k1 is narrower compared with samples k1-k5, and with prolonging the calcination time, the distributions become wider. The average particle size of samples k1-k5 is shown in Fig.5. With prolonging the calcination time, the average particle size becomes large fast.

Fig.4 Size distribution of 2% WO3-TiO2 catalysts with oxygen vacancies

Fig.5 Dependence of average particle diameter on heating time of 2% WO3-TiO2 with oxygen vacancies
XPS spectra of element W on the surface of samples k0, k3 and k5 are shown in Fig.6. For sample k0, there is only one kind of valence for W element and it is in the valence of +6. The band energies of W 4f7/2 and 4f5/2 are 35.900 eV and 37.590 eV, respectively. But the third peak “t” is for Ti 3p in TiO2, and the band energy is 37.00 eV. All these present that W element on the surface of WO3/TiO2 which is not thermally treated in H2 atmosphere is only in valence of +6. However, for sample k3, there are two kinds of valences for W element on the surface of WO3/TiO2, +6 and +4. The band energies of W4+ 4f7/2, W4+ 4f5/2 are 33.483 eV and 35.653 eV, respectively. For sample k5, there is only one kind of valence for W element and it is in the valence of +4, which shows that after 9 h calcination WO3 has changed to WO2 entirely. This is in agreement with the result of XRD.

Fig.6 XPS patterns of W element in 2% WO3-TiO2 catalysts with oxygen vacancies: (a) k0; (b) k3; (c) k5
XPS spectra of element Ti on the surface of samples k1 and k5 are shown in Fig.7. The band energies of Ti4+ 2p1/2, Ti4+ 2p3/2, Ti3+ 2p1/2 and Ti3+ 2p3/2 are 465.40, 459.500, 464.51 and 458.940 eV, respectively. For sample k1 the peak areas of Ti4+ 2p1/2 and Ti4+ 2p3/2 are 1 702.863 and 5 083.813, and those of Ti3+ 2p1/2 and Ti3+ 2p3/2 are 1 329.947 and 2 447.097, respectively. According to the peak areas, the contents, of Ti4+ and Ti3+ are 64.25% and 35.75%, respectively. Compared with the XPS spectra of the sample k1, the position of each peak in sample k5 is unchanged, but the peak areas are changed: 1 271.861, 4 203.676, 1 781.036 and2 600.000 for Ti4+ 2p1/2, Ti4+ 2p3/2, Ti3+ 2p1/2 and Ti3+ 2p3/2, respectively. Thus, the contents of Ti4+ and Ti3+ change to 55.55% and 44.45%, respectively. So, with prolonging the calcination time, the Ti3+ content increases, which means that the amount of oxygen vacancies increases with increasing the Ti3+ content.
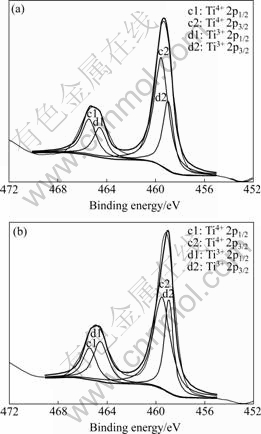
Fig.7 XPS patterns of Ti element in 2% WO3-TiO2 catalysts with oxygen vacancies: (a) k1; (b) k5
3.2 Photocatalytic activity of catalyst
The photocatalytic activities of the 2% WO3-TiO2 catalysts with oxygen vacancies were studied through the splitting of water for O2 evolution under UV-light irradiation. The results are presented in Fig.8. The results show that the subsequence for O2 evolution of the catalysts are as follows: k3>k2>k4>k5>k1>k0, under 12 h irradiation. The maximum rate for O2 evolution is 667 ?mol/(L・h) (k3). For k1 and k2, the subsequence of O2 evolution has a good agreement with their oxygen vacancy concentration and the absorption ability within 420-700 nm. For k4 and k5, with the increase of the concentration of the oxygen vacancies, the absorbing ability becomes stronger, and the O2 evolution rates become slower.
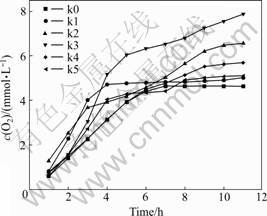
Fig.8 Photocatalytic activity for O2 evolution of splitting water of 2% WO3-TiO2 catalysts with oxygen vacancies
When the surface of the semiconductor is irradiated by the light whose energy is larger than the band gap of the semiconductor, electrons will obtain enough energy to jump onto the conduction band and become free electrons named photoelectrons. Thus, holes are left in the valence band[20]. Generally, The photocatalytic activity of photocatalyst is determined by its ability of light absorbing, the efficiency of separation between photoelectrons and holes, and the transfer rate of charge carriers[21]. With prolonging the calcination time, the particle size is enlarged, and the BET is decreased. The decrease of BET leads to the weakness of the absorbing ability and the photocatalytic reaction for H2 or O2 evolution in this triphase reactor of solid-liquid-gas. The photo-redox reaction proceeds on the surface of the catalysts. So, on one hand the decrease of the BET of catalysts leads to its lower O2 evolution rate. On the other hand, when the calcination time is prolonged, the oxygen vacancy concentration increases. Appropriate oxygen vacancies can obviously improve the efficiency of the separation of electrons and holes and the absorbing ability for Vis-light, so as to improve the photocatalytic activity of the photo-catalysts[19]. However, too much oxygen vacancies could bond too much charge, leading to the decrease of the efficiency of electron-hole separation and photocatalytic activity.
4 Conclusions
1) The 2% WO3-TiO2 catalysts with oxygen vacancies were prepared using H2 atmosphere calcined at 873 K. The XRD patterns show that some distinct peaks for WO2 can be observed after being thermally treated for 2 h (k1), and peaks for WO3 disappear after being thermally treated for 9 h (k5).
2) When the calcination time is prolonged, the particle size is enlarged. The EPR results show that very weak signals of Ti3+ and OV are present after being thermally treated for 2 h, and strong signals of Ti3+ and OV are present after being thermally treated for 5 h. The UV-Vis spectra of the catalysts show that the samples have the same absorption ability of UV-light and different absorption ability of visible light. Longer time calcination means better absorption of visible light.
3) From the XPS spectra, the content of Ti3+ increases with the calcination time.
4) Using Fe3+ as the electron acceptor, the maximum rate for O2 evolution is 667 ?mol/(L・h) under UV irradiation in 12 h.
References
[1] TERUHISA O, FUMIHIRO T, KAN F, SHINOBU I, MICHIO M. Photocatalytic oxidation of water by visible light using ruthenium-doped titanium dioxide power [J]. Photochemistry and Photobiology A: Chem, 1999, 127: 107-110.
[2] KIM J C, CHOI J K, LEE Y B, HONG J H, LEE J I, YANG J W, HUR N H. Enhanced photocatalytic activity in composites of TiO2 nanotubes and CdS nanoparticles [J]. The Chemical Communication, 2006, 46: 5024-5026.
[3] JING L Q, FU H G, WANG B Q, WANG D J, XIN B F, LI S D, SUN J Z. Effects of Sn dopant on the photoinduced charge property and photocatalytic activity of TiO2 nanoparticles [J]. Applied Catalysis B: Environmental, 2006, 62(3): 282-291.
[4] MIN S X, WANG F. Synthesis and photocatalytic properties of nanometer TiO2/conjugated polymer complex materials [J]. Chemical Bulletin, 2008, 4: 297-302.
[5] ZHANG Qi, LI Xin-jun, LI Fang-bai. Effect of preparation process of WO3/TiO2 films on photo-catalytic activity under visible light [J]. Trans Nonferrous Met Soc China, 2002, 12(6): 1299-1303.
[6] TONG Hai-xia, CHEN Qi-yuan, YIN Zhou-lan, HU Hui-ping, LI Jie, ZHAO Li. Preparation of TiO2 photocatalyst coated with WO3 for O2 evolution [J]. Trans Nonferrous Met Soc China, 2008, 18(4): 682-687.
[7] GAO You-liang, CHEN Qi-yuan, YIN Zhou-lan, ZHOU Jian-liang, LI Jie. Photocatalytic activity for O2 evolution of WO3 prepared through pyrolysis of ammonium paratungstate under O2/Ar atmosphere [J]. Trans Nonferrous Met Soc China, 2006, 16(5): 904-908.
[8] HARRIS L A, SCHUMACHER R. The influence of preparation on semiconducting rutile (TiO2) [J]. J Electrochem Soc: Solid-state Sci Technol, 1980, 127(5): 1186-1188
[9] HELLER A, DEGANI Y, JOHNSON D W, GALLAGHER P K.Controlled suppression and enhancement of the photoactivity of titanium-dioxide (rutile) pigment [J]. J Phys Chem, 1987, 91(23): 5987-5991.
[10] CHEN Y X, WEI Z B, CHEN Y X, LIN H X, HONG Z P, LIU H Q, DONG Y L, YU C Y, LI W Z. Metal-semiconductor catalyst-photocatalytic and electrochemical behavior of Pt-TiO2 for the water gas shift reaction [J]. J Mol Catal, 1983, 21(10): 275-289.
[11] HOWE R F, GR?TZEL M. Electron-paramagnetic-resobservation of trapped electrons in colloidal TiO2 [J]. J Phys Chem, 1985, 89(21): 4495-4499.
[12] QIN D, CHANG W D, CHEN Y, ZHOU J L, CHEN Y Q, GONG M C. Dynamic ESR study of oxygen chemisorption on TiO2-based catalysts [J]. J Catal, 1993, 142(2): 719-724.
[13] REKOSKE J E, BARTEAU M A. Isothermal reducing kinetics of titanium dioxide-based materials [J]. J Phys Chem B, 1997, 101(7): 1113-1124.
[14] LIU H, MA H T, LI X Z, LI W Z, WU M, BAO X H. The enhancement of TiO2 photocatalytic activity by hydrogen thermal treatment [J]. Chemosphere, 2003, 50: 39-46.
[15] JIANG Y H, SUN Y M, ZHAO C X, WU M, YIN H B, CHEN K M. Effects of polyatomic acids as modifiers on hydrothermal synthesize of rutile TiO2 nanorods [J]. New Chemical Materials, 2007, 35(4): 28-30.
[16] TORIMOTO T, FOX III R J, FOX M A. Photoelectro-chemical doping of TiO2 particles and the effect of charge carrier density on the photoactivity of microporous semiconductor electrode films [J]. J Electrochem Soc, 1996, 143(11): 3712-3717.
[17] OH S M, ISHIGAKI T. Preparation of pure rutile and anatase TiO2 nanopowders using RF thermal plasma [J]. Thin Solid Films, 2004, 457: 186-191.
[18] XU Yu-long. Semiconductor basis on oxides and compounds [M]. Xi’an: Xi’an University of Electronic Science and Technology Press, 1991: 49. (in Chinese)
[19] CHEN Q Y, TONG H X, YIN Z L, HU H P, LI J, LIU L L. Preparation, characterization and photo-catalytic behavior of TiO2 catalysts with oxygen vacancies [J]. Acta Phys-Chim Sin, 2007, 23(12): 1917-1921.
[20] HAN Wei-ping. Catalytic chemistry introduction [M]. Beijing: Science Press, 2003: 272. (in Chinese)
[21] ZHANG Q, LI X J. Investigation on visible-light activity of WOx-TiO2 photocatalyst [J]. Phys Chem Trans, 2004, 20(5): 507-511.
Foundation item: Project(08JJ3022) supported by the Natural Science Foundation of Hunan Province, China; Project(K-081025) supported by the Photocatalytic Key Laboratory of Fujian Province, China
Corresponding author: TONG Hai-xia; Tel: +86-731-85258733; E-mail: tonghaixia@126.com
DOI: 10.1016/S1003-6326(09)60056-X
(Edited by YANG Bing)

