稀有金属 2010,34(01),63-67
Ir1-xRuxO2/Ti析氧阳极的电催化活性研究
李山梅 叶锋 魏浩杰 侯淼淼 王同涛 王新东
北京科技大学冶金学院物理化学系
摘 要:
以氯铱酸为主要的前驱体,采用加热分解法制备了不同Ru含量的Ir1-xRuxO2/Ti析氧阳极,并采用扫描电镜,循环伏安,恒电流电解,线性极化等测试手段对电极进行表征和测试。电化学测试结果表明当Ir,Ru的摩尔比为3∶2时,500mA.cm-2恒电流水解电位最低,为1.4V(vs.SCE);循环伏安测试表明,Ir0.6Ru0.4O2/Ti的伏安电量亦达到最高,达到1271mC.cm-2,析氧电催化活性点最多。SEM表面形貌进一步证实了Ru的加入使得电极的表面的多孔结构更加明显,当Ir,Ru摩尔比为3∶2时,电极表面颗粒最小,孔隙率最高,亦表明该电极的电化学活性表面积最大,电催化活性最高。
关键词:
Ir1-xRuxO2/Ti;电催化活性;多孔性;
中图分类号: TF351
作者简介:李山梅(1984-),女,河北石家庄人,硕士;研究方向:质子交换膜水电解;王新东(E-mail:echem@ustb.edu.cn);
收稿日期:2009-05-27
基金:国家863计划(2007AA05V150)资助项目;
Electrocatalytic Activity of Ir1-xRuxO2/Ti Anodes for Oxygen Evolution
Abstract:
Ir1-xRuxO2/Ti anodes with different content of Ru prepared by thermo-decomposition using ethanol and isopropanol mixed solution of chloroiridic acid (H2IrCl6) and ruthenium chloride (RuCl3) were characterized by SEM and investigated by cyclic voltammetry,charonopotentiometry,liner polarization. The electrochemical tests indicated that the potential with the electrolysis current of 500 mA・cm-2 was only 1.4 V (vs.SCE) corresponding to the electrode with metal content of 40% Ru; cyclic voltammetry test indicated that q* of Ir0.6Ru0.4O2/Ti anode reached the maximum value of 1271 mC・cm-2,which existed a maximum of active points on the surface of the anode. The pictures of SEM also showed the most significant porous morphology of crystallite grains packing and cracks on the surface of anode corresponding to the electrode with metal content of 40% Ru,which coincided with the electrochemical test result.
Keyword:
Ir1-xRuxO2/Ti; electrocatalytic activity; porous morphology;
Received: 2009-05-27
氢能作为一种清洁,高效的能量形式越来越受到世界各国的广泛关注和研究,质子交换膜(PEM)电解水制氢具有对环境无污染,无温室气体排放等优点,具有良好的发展前景。然而质子交换膜电解水制氢由于能耗高,效率低制约了其发展,主要原因在于阳极析氧电位过高。因此氧阳极是制约质子交换膜电解水技术发展的关键材料之一。由于目前所使用的固体电解质膜为全氟磺酸型质子交换膜,当PEM电解池充满水时,质子交换膜具有很强的酸性,且析氧电位较高(氧气析出的标准电位为1.29 V vs.RHE),这使得电解池的阳极将处于腐蚀性极强的环境中。这种恶劣的运行环境,使得析氧催化剂的选择几乎完全局限在Pt系贵金属及其合金上。
Marshall A等[1]用Adams法[2]制备的IrO2显示了良好的析氧电催化活性。根据对电位控制理论[3,4],只有当阳极电位高于金属/氧化物或低于氧化物/高价氧化物对的标准电极电位时,氧化物表面的析氧过程才会发生,即控制对的标准电极电位越低,氧化物的析氧活性越大。Ir2O3/IrO2的标准电位在铂族金属氧化物中是最低的[5],说明了IrO2在酸性介质中具有很高的析氧电催化活性。然而当电流密度较高时,其寿命便难以满足使用要求。Marshall A等后来的研究[6]表明IrO2-RuO2两元氧化物体系的析氧催化活性更高,因为亲氧元素Ru的加入,可以有效改善IrO2的结构,使析氧催化活性更高,稳定性更好,但具体针对不同Ru含量的氧电极的基础研究尚少。本文采用加热分解法制备了不同Ir,Ru摩尔比的氧电极的表征与性能测试,寻找最佳的Ir,Ru摩尔比并分析讨论不同配比下的氧电极析氧活性机制。
1 实验
1.1 电极的制备
钛板的预处理:将纯钛板进行喷砂处理,而后置于洗衣粉和无水碳酸钠混合溶液中加热至85~90℃煮沸15~60 min,除去表面油迹,用毛刷洗去残渍,去离子水冲洗干净,烘干;然后浸于30%的HCl溶液,加热到90℃蚀刻2 h后,迅速用大量去离子水冲洗钛基体表面残液,烘干后置于乙醇中浸泡备用。
催化剂涂层阳极的制备:配置金属离子浓度为0.1 mol L-1的氯铱酸和三氯化钌催化剂前驱体溶液,溶剂为乙醇和正丙醇的混合溶液。其中铱钌原子比分别为1∶0,4∶1,3∶2,2∶3,1∶4。用微量移液管移取不同Ir,Ru摩尔比的催化剂前躯体溶液,滴涂于钛板上并让其自然扩散至整个催化剂区域,先于烘箱中90℃烘干10 min,取出后置于管式电阻炉中450℃烧结10 min后,取出空冷,重复以上过程10次,最后于电阻炉中450℃退火处理1 h得到5种不同配比的析氧阳极(电极表面催化剂表观面积为1 cm2)。
1.2 物理及电化学表征
采用JSM-6480LV型扫描电镜测试仪进行表面形貌的测试。
采用传统的三电极体系,以析氧阳极为工作电极,Pt片为对电极,饱和甘汞电极为参比电极,所用的电解质溶液为0.1 mol L-1的H2SO4溶液,工作温度为25±1℃。循环伏安,线性极化,恒电流电解等电化学测试由VMP(美国Ametek公司)电化学测试仪完成。
2 结果与讨论
2.1 表面结构形貌
上述加热分解方法制备的5种不同Ru含量的催化剂涂层电极的扫描电镜表面形貌如图1所示(放大倍率为2000),5种不同的催化剂表面均有不同程度的裂缝。单一IrO2的表面呈分裂开来的许多小平面状,最为平滑;Ir0.8Ru0.2O2/Ti电极表面呈开裂的干泥状,裂缝较多亦较平整;而Ir0.6Ru0.4O2/Ti电极表面由分布均匀的催化剂颗粒聚集,颗粒大小约为几个微米,颗粒聚集与裂缝的存在使得该电极呈现多孔状结构;Ir0.4Ru0.6O2/Ti表面亦呈颗粒聚集状,但颗粒尺寸较大,分布亦不均匀,Ir0.2Ru0.8O2/Ti表面呈颗粒堆积的泥状,裂缝很少,多孔结构与图1(c),(d)相比较不明显。从SEM图像可以明显看出,当Ir,Ru摩尔比为3∶2时,催化剂表面多孔结构最明显。

2 0.8 0. 2 2 0. 6 0. 4 2 0. 4 0. 6 2 0.8 0. 2 2Fig.1 SEM of IrO2/Ti,Ir0.8Ru0.2O2/Ti,Ir0.6Ru0.4O2/Ti,Ir0.4Ru0.6O2/Ti,Ir0.8Ru0.2O2/Ti
(a)IrO2/Ti;(b)Ir0.8Ru0.2O2/Ti;(c)Ir0.6Ru0.4O2/Ti;(d)Ir0.4Ru0.6O2/Ti;(e)Ir0.2Ru0.8O2/Ti
2.2 析氧性能表征
为了表征不同Ru含量的Ir1-xRuxO2电极的电催化性能,对各电极进行了的线性极化测试和500 mA cm-2恒电流测试,测试结果如图2,3所示。从图2曲线可以看出,各电极含Ru量虽然不同,但开始析氧的电位都维持约1.16 V的位置。当析氧电流小于200 mA cm-2时,Ir0.6Ru0.4O2/Ti电极的析氧电位并非最低,与Ir0.8Ru0.2O2/Ti及Ir0.4Ru0.6O2/Ti电极的析氧电位相近;然而当电流逐渐增大时,Ir0.6Ru0.4O2/Ti电极的极化电位增长最为缓慢。当极化电位为1.5 V时,Ir0.6Ru0.4O2/Ti电极析氧电流达到最大,约为800 mA cm-2。可见Ir0.6Ru0.4O2/Ti电极表面参与反应的电催化活性点的数目最多,催化剂活性最大。从Ir0.6Ru0.4O2/Ti电极的SEM图中可以看出,该电极的表面由于催化剂颗粒聚集以及裂缝的存在,使得与溶液接触的表面积最大,电化学反应的真实面积亦最大,因此1.5 V时析氧电流远远大于其他电极。
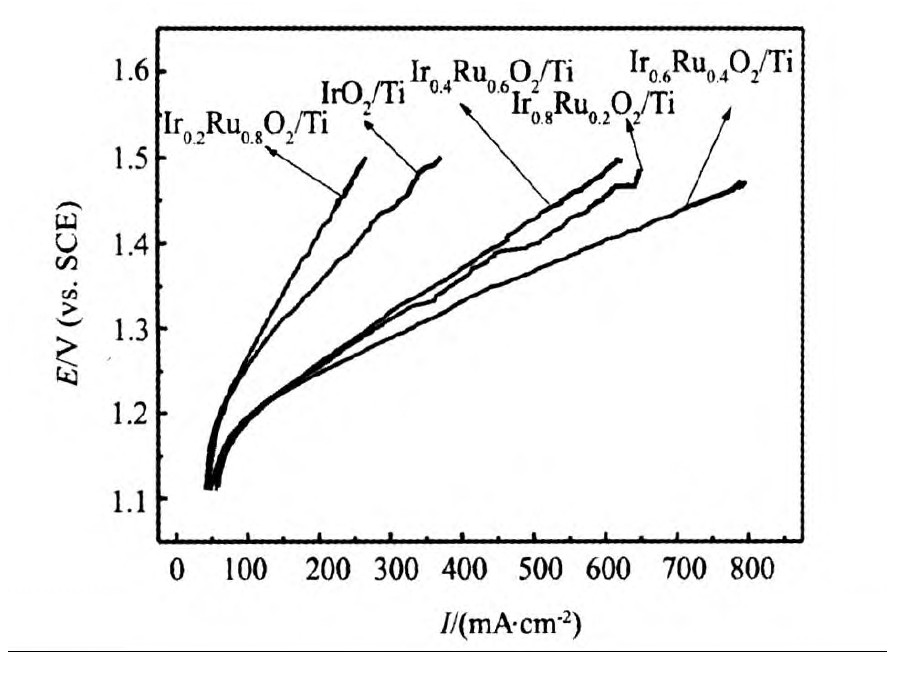
图2 Ir1-xRuxO2/Ti在0.1 mol L-1 H2SO4溶液中的线性极化曲线Fig.2 Liner polarization curve of Ir1-xRuxO2/Ti in 0.1 mol L-1H2SO4
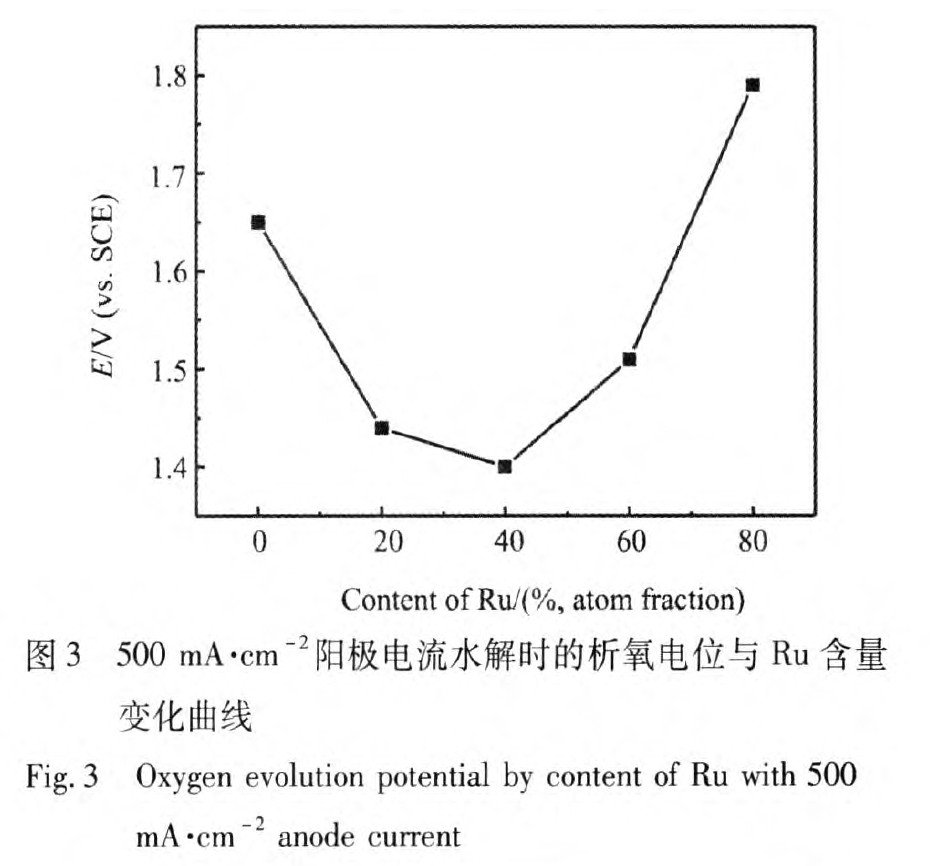
从图3中可以看出,当通以500 mA cm-2析氧电流时,析氧电位随Ru含量的增大先降低后升高,Ir0.6Ru0.4O2/Ti电极的析氧电位最低为1.4 V,这一结果与以上线性极化的试验结果相一致。随着氧电极Ru含量的进一步增大,电极的析氧电位急剧上升,这可能是由于随着Ru含量过大时,催化剂层与基体之间的结合力变差,催化剂的表面状态不稳定,随着电解反应的进行,电催化活性点的数目越来越少,从而使得Ir0.2Ru0.8O2/Ti电极的析氧活性最低。
为了对电极的析氧电催化活性进行进一步的探究对各电极进行了循环伏安测试,图4为以-0.24~1.26 V(vs.SCE)为电位区间,扫描速度为100 mV s-1时不同Ir,Ru摩尔比催化剂的循环伏安曲线,从图4中可以看出Ir0.6Ru0.4O2/Ti电极的电化学窗口最宽,IrO2与Ir0.8Ru0.2O2/Ti电极的次之,Ir0.4Ru0.6O2/Ti电极再次之,Ir0.2Ru0.8O2/Ti电极的窗口最窄。在相同的电极电位下,析氧电流密度随着电极Ru含量的增加先增大后减小,而Ir0.6Ru0.4O2/Ti电极的析氧性能最佳。
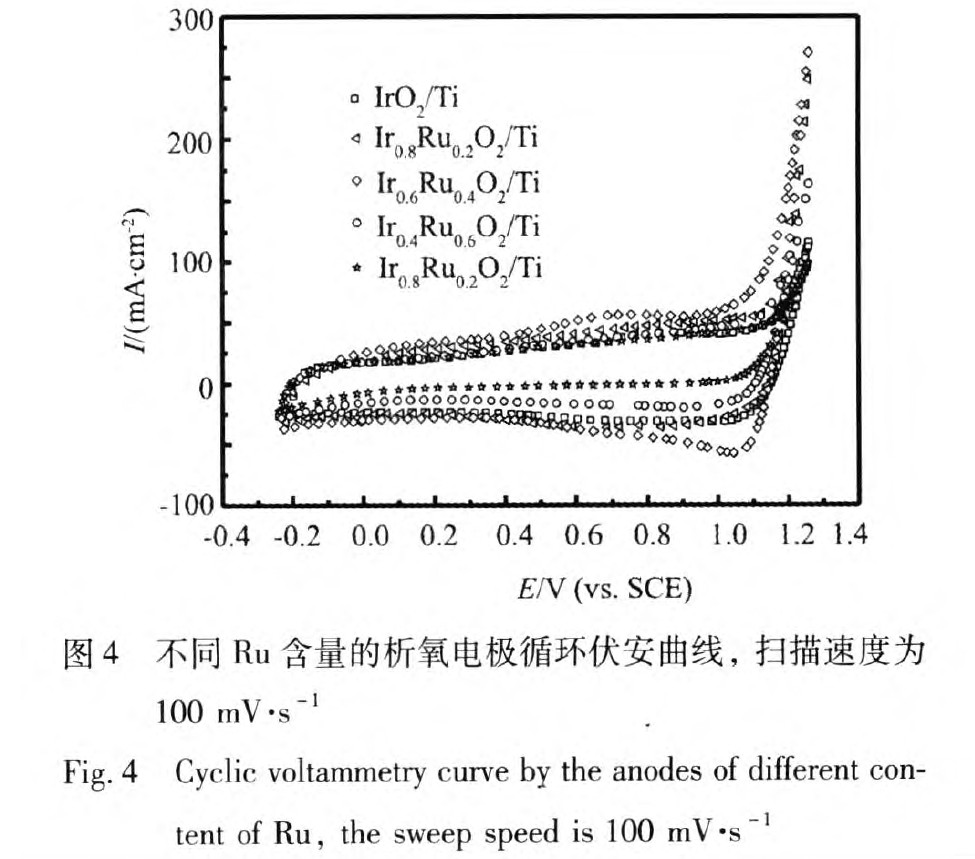
研究表明[6,7]由循环伏安曲线得到的伏安电量q*可以用来表征催化剂电极表面的电化学反应活性点数,从循环伏安图中可以看出,析氧反应开始的电位大约在1.16 V(vs.SCE)左右,因此选择伏安电量的积分区间为0 V(vs.SCE)-1.16 V(vs.SCE)[8],该电量的计算由软件EClab积分计算出,图5所示为不同Ir,Ru摩尔比的电极的伏安电量q*曲线。
研究表明[9],阳极表面的催化过程是由电极表面的电催化活性点数目的多少决定的。电化学活性点越多,电极表面参与电催化反应的活性面积就越大,电极的电催化性能越好。从图5中可以看出IrO2电极的q*最小,约800 mC cm-2。随着Ru含量的增大,电极的伏安电量变化呈先增大后减小的趋势,当Ir,Ru摩尔比为3∶2时,电极的q*达到最大,为1271 mC cm-2,表明Ir0.6Ru0.4O2/Ti电极参与电催化反应活性点最多,电催化性能最好。从SEM上亦可以看出由于Ru原子的加入,催化剂的表面形貌得到改善,当Ir,Ru摩尔比为3∶2时,电极表面的催化剂颗粒粒径最细小,表面孔隙率最大,电化学活性面积最大,因此活性点数目最多。催化剂颗粒的边缘处与裂缝处[10,11]都是析氧催化反应发生的主要界面,裂纹和催化剂颗粒聚集的存在增大了析氧反应的真实表面积,然而裂纹的存在使得电解液向电极基体的渗透变的严重,电极的腐蚀随之加剧,从而导致了图1(a)所示电极在大电流下电解时的不良性能。
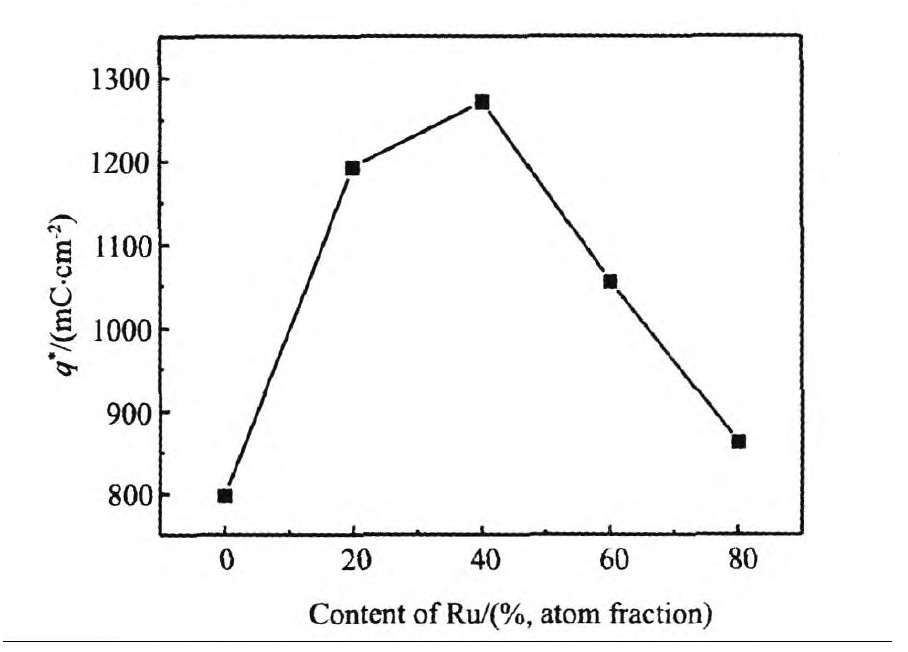
图5 不同电极的q*随Ru含量变化曲线,扫描速度为100mV s-1Fig.5 Curve of q*by content of Ru,the sweep speed is100 mV s-1
3 结论
Ir1-xRuxO2/Ti电极的电催化性能随着Ru含量的增大先升高后降低,当Ir,Ru的摩尔比为3∶2时,电极q*最大,达到1271,500 mA cm-2恒电流水解电位最低,为1.4 V(vs.SCE),Ir0.6Ru0.4O2/Ti电极的电催化活性最高。SEM图也说明了当Ir,Ru摩尔比为3∶2时,电极表面催化剂颗粒最细小,分布最均匀,电极的活性表面积最大,电极表面活性点最多,电极的电催化活性最高。
参考文献
[1] Marshall A,Bφrresen B,Hagen G,Tsypkin M,Tunold R.Electrochemical characterisation of IrxSn1-xO2powders as oxygen evolution electrocatalysts[J].Electrochemica.Acta,2006,51(15):3161.
[2] Adams R,Shriner R L.Simplifivstion of the gattermann synthe-sis of hydroxyl baldheads[J].American Chemical Society,1923,45:2171.
[3] Tseung A C,Jasem S.Oxygen evolution on semiconducting ox-ides[J].Electrohcimica.Acta,1977,22(12):31.
[4] Rasiyah P,Tseung A C.The role of the lowmetal oxide/high metal oxide couple in oxygen evolution reaction[J].Electrochim-ica.Soc.,1984,131(4):803.
[5] Angelinetta C,Trasatti S,Atanasoska Lj D,Minevski Z S,Ata-nasoski R T.Effect of preparation on the surface and electrocat-alytic properties of RuO2+IrO2mixed oxide[J].Materials Chemistry and Physics,1989,22(1):231.
[6] Marshall A,Bφrresen B,Hagen G,Tsypkin M,Tunold R.Hy-drogen production by advanced proton exchange membrane(PEM)waterelectrolysers-reduced energy consumption by im-proved electrocatalysis[J].Energy,2007,32(4):431.
[7] Ye Zhiguo,Meng Huimin,Chen Dong,Yu Hongying,Huan Zh-ishan,Wang Xudong,Sun Dongbai.Structure and characteris-tics of Ti/IrO2(x)+MnO2(1-x)anode for oxygen evolution[J].Solid State Science,2008,10(4):346.
[8] Krishtalik L I.Kinetics and mechanism of anodic chlorine andoxygen evolution reactions on transition metal oxide electrodes[J].Electrochim.Acta,1981,26(3):329.
[9] Xu Likun,Xin Yonglei,Wang Juntao.A comparative study on IrO2-Ta2O5coated titanium electrodes prepared with different methods[J].Electrochimica Acta,2009,54(6):1820.
[10] Xu L K,Scantlebury J D.A study on the deactivation of anIrO2-Ta2O5coated titanium anode[J].Corrosion Science,2003,45(12):2729.
[11] Yao Shudian,Shen Jianian.Variation of morphology and com-position of IrO2+Ta2O5coated titanium anode in OER[J].Pre-cious Metals,2006,27:4.

