网络首发时间: 2018-02-06 17:48
稀有金属 2018,42(10),1068-1076 DOI:10.13373/j.cnki.cjrm.xy17090012
H2O对不同状态Pd催化剂上低浓度CO催化氧化性能的影响
吴彦町 张艳慧 王丽 郭杨龙 詹望成 郭耘
华东理工大学工业催化研究所结构可控先进材料教育部重点实验室
摘 要:
采用不同的Pd前驱体, 通过浸渍法制备了系列Al2O3负载Pd催化剂, 研究表明:Pd的化学状态、卤素离子的引入等不仅可直接影响催化剂的氧化活性, 还可影响气氛中H2O的作用。当Pd以氧化态存在时, 以Pd (NO3) 2的HBr溶液为前驱体所制备得到的BrC催化剂具有最高的反应活性, 而以PdCl2的HCl溶液为前驱体所得ClC催化剂的活性最低;经H2还原后, 催化剂的活性均有显著的提高, 其中以Pd (NO3) 2为前驱体所得CH催化剂表现出最高的CO氧化活性, 另外两种催化剂活性相差不大。同时对于还原态催化剂, 气氛中的H2O可抑制CH表面CO的吸附, 并有利于O2的吸附和活化, 从而显著提高了CO的氧化活性;但Cl和Br引入后, H2O的存在对CO吸附和活性影响不明显。当Pd以氧化态形式存在时, H2O的引入可促进BrC和C催化剂上的CO氧化, 其中BrC催化剂上的促进作用最为明显, 但却抑制了以PdCl2的HCl溶液为前驱体所得ClC催化剂上的CO氧化。
关键词:
CO;催化氧化;Pd催化剂;水的影响;
中图分类号: O643.36
作者简介:吴彦町 (1991-) , 男, 江苏镇江人, 硕士, 研究方向:环境催化;E-mail:wuyanding0950@163.com;;*王丽, 副教授;电话:021-64253703;E-mail:wangli@ecust.edu.cn;
收稿日期:2017-09-06
基金:国家重点基础研究发展计划项目 (2016YFC0204300, 2013CB933201);国家自然科学基金项目 (21571061);上海市教育委员会曙光计划项目 (12SG29) 资助;
Role of H2O in Low Concentration CO Oxidation on Pd Catalysts with Different States
Wu YANDing Zhang Yanhui Wang Li Guo Yanglong Zhan Wangcheng Guo Yun
Key Laboratory for Advanced Materials ( Ministry of Education) , Research Institute of Industrial Catalysis, East China University of Science and Technology
Abstract:
Pd/Al2O3 catalysts were prepared by impregnation method using different precursors of Pd, the activities of CO oxidation and the roles of H2O in the reaction were investigated. The results showed the chemical states of Pd and the exit of halogen ions ( Cl/Br) could directly affect the activities of CO oxidation and the role of H2O. For the Pd with high chemical valence, Br C catalyst prepared by using HBr solution of Pd ( NO3) 2 as precursor showed the highest activity for CO oxidation. While Cl C catalyst prepared by using HCl solution of PdCl2 as precursor showed the lowest activity. After the reduction by H2, the activities of catalysts were promoted significantly. Among them, CH catalyst prepared by using Pd ( NO3) 2 solution as precursor behaved the highest activity. The activity of other catalysts showed no difference. Meanwhile, the presence of H2O could block CO adsorption on the surface of CH catalyst and enhance the adsorption and activation of O2, which could accelerate CO oxidation significantly. However, the presence of Cl/Br did not bring observable effects on the CO adsorption and oxidation. For the Pd with high chemical valence, H2O in the feed gas could promote CO oxidation on Br C and C catalyst. Among them, Br C showed the highest promotion. But block the CO oxidation on the Cl C catalyst prepared by using HCl solution of PdCl2 as precursor.
Keyword:
CO; catalyst oxidation; Pd catalyst; effects of H2O;
Received: 2017-09-06
CO催化氧化不仅具有重要的应用价值, 同时也是多相催化中最常用的模型反应, 用于研究催化剂的表面性质。CO氧化催化剂按其组成, 大体上可分为两种, 一种是非贵金属体系, 另外一种是贵金属体系。其中, 非贵金属体系催化剂主要是指过渡金属 (复合) 氧化物[1,2,3,4], 包括单组分金属氧化物, 例如氧化钴 (Co3O4) 和氧化锰 (MnOx) 催化剂, 以及多组分的复合氧化物催化剂体系。其中最典型代表是已经商品化生产的Hopcalite催化剂。贵金属催化剂体系最典型代表是负载型金 (Au) [5,6,7,8], 铂 (Pt) [9,10,11,12]和钯 (Pd) [13,14,15,16]催化剂, 其中Pt和Pd催化剂因具有良好的催化活性和稳定性, 已经在汽车尾气净化等领域得到了广泛应用。
在CO氧化的实际应用过程中, 如汽车尾气净化、隧道空气污染物净化等, 反应气氛中不可避免有大量水的存在。理论计算的结果表明, 由于金属表面所吸附的OH与CO形成过渡态的势能面比O与CO形成过渡态的势能面更为平坦, 因此水的引入可促进金属表面CO的催化氧化[17]。
本文主要是针对低浓度CO的催化氧化, 制备了具有不同Pd化学态的负载型Pd/Al2O3催化剂, 研究了气氛中水对具有不同化学状态的Pd催化剂上CO催化氧化性能的影响。
1 实验
1.1 催化剂制备
催化剂制备以Pd (NO3) 2, PdCl2等为前驱体, 以去离子水、10%的盐酸或氢溴酸作为溶剂, 按照固定比例配制出理论Pd负载量为1%的溶液, 采用等体积浸渍法将20~40目的Al2O3载体浸渍于上述溶液中24 h。浸渍完成后, 经旋蒸去除多余溶液, 110℃干燥、马弗炉550℃下焙烧3 h, 得到成品催化剂。催化剂的标识如表1所示。
1.2 催化剂评价
CO氧化活性评价使用的评价仪器为福立9790型气相色谱, 在直型固定床石英反应管中进行的, 样品颗粒大小为20~40目, 原料气浓度为为100×10-6CO-20% (体积分数) O2-N2配平。在评价过程中, 混合气体的总流速为50 ml・min-1, 空速为60000 h-1。
表1 样品标记一览表Table 1 List of sample tags 下载原图
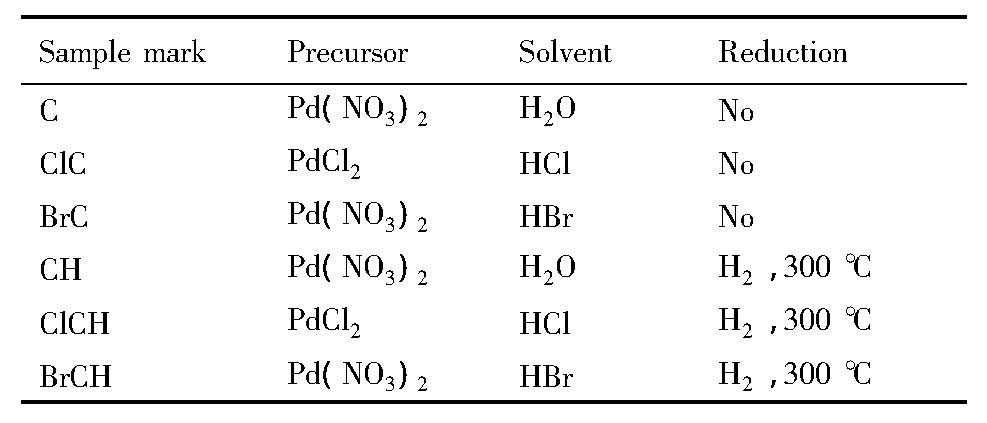
表1 样品标记一览表Table 1 List of sample tags
1.3 催化剂表征
1.3.1 X射线粉末衍射 (XRD)
X射线衍射分析 (XRD) 使用Bruker D8衍射仪进行检测, Cu Kα靶作为入射光源, λ=0.154056 nm, 管压为40 k V, 管流为40 m A, 扫描范围为10°~80°, 扫描速率为6 (°) ・min-1。
使用药匙将样品装入样品槽内, 然后使用载玻片将样品压实, 表面压平整进行扫描。样品的晶粒大小Scherrer公式 (1) 进行计算。

式中, Dp为粒子直径, k为Scherrer常数 (0.89) , λ为入射X光波长 (0.15406 nm) , θ为衍射角 (°) , β1/2为衍射峰的半高峰宽 (rad) 。
1.3.2 原位红外漫反射 (In Situ-DRIFTS)
原位红外漫反射 (in situ-DRIFTS) 检测使用的是Nicolet Nexus 670光谱仪进行表征, 配备了MCT检测器, Zn Se样品池及温度控制仪, 检测器用液氮冷却, 分辨率为8 cm-1, 扫描次数为32次。40 mg样品经研磨后放入样品池用KBr窗口罩封, 先用20 ml・min-1H2在200℃下预处理0.5 h, 然后再Ar气氛中 (30ml・min-1) 冷却至60℃, 采集样品背景。随后将CO气氛通入样品再用Ar气吹扫后记录光谱。
2 结果与讨论
2.1 氧化态Pd催化剂上CO催化氧化
2.1.1 氧化态Pd催化剂的XRD表征
对3种催化剂进行了XRD表征, 结果如图1所示。
从图1中可以看出3种样品的XRD图均显现出Al2O3特征峰, 没有检测到Pd O物种特征峰, 这表明PdO物种在3种催化剂上以高分散的形式存在。
2.1.2 氧化态Pd催化剂的CO催化氧化性能
当Pd以氧化态形式存在时, 催化剂上CO氧化性能的评价结果列于图2。
从图2中可以看出, 以Pd (NO3) 2的水溶液为前驱体所制备的催化剂C上, 在低温段CO转化率随温度的升高快速增加。当反应温度超过60℃后, CO转化率随温度上升的提高幅度变缓, 160℃时CO转化率趋于60%。反应气氛中引入水, CO的氧化活性在低温段稍许降低;但当温度达到60℃以上之, 活性显著提高。
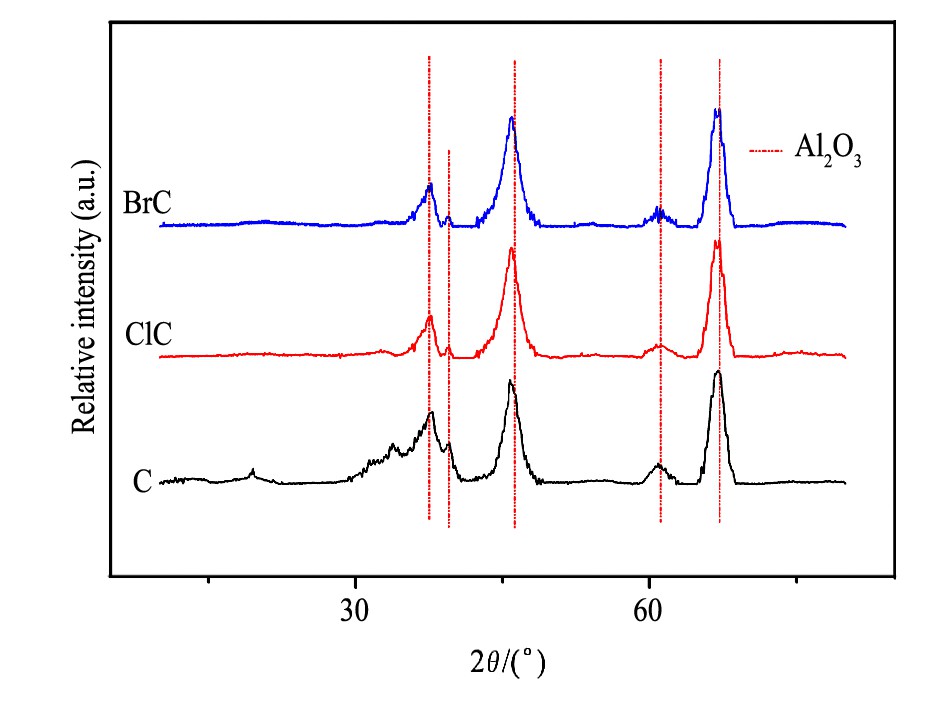
图1 含不同卤素氧化态样品的XRD图Fig.1 XRD patterns of oxidized samples containing different halogen
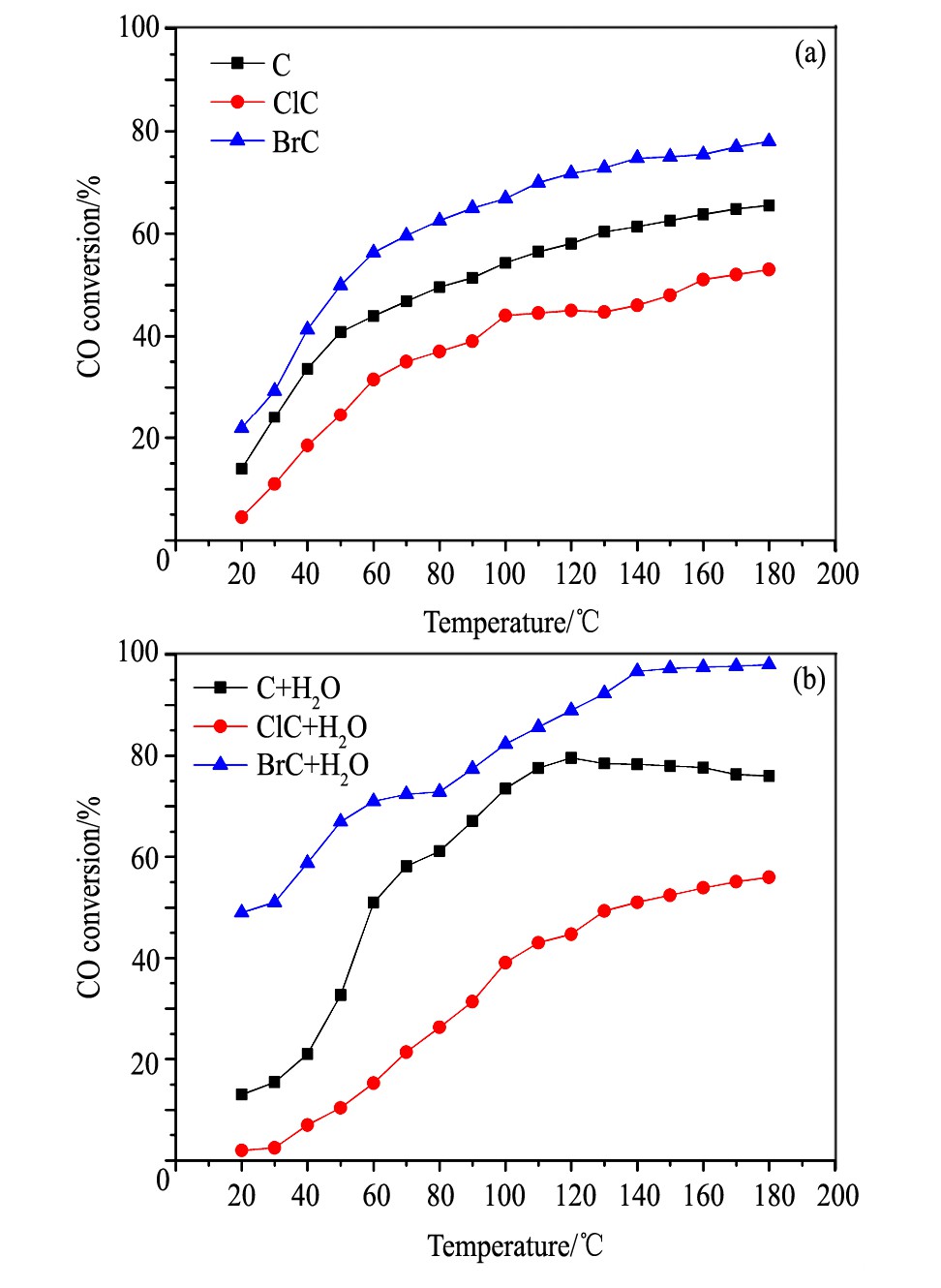
图2 含不同卤素氧化态样品的CO氧化活性Fig.2 CO oxidation activity of oxidized samples containing dif-ferent halogen before (a) and after (b) water vapor ac-cessing
与催化剂C相比, 以PdCl2的HCl溶液为前驱体所制备的催化剂ClC的活性较低。在无水条件下, 反应温度达到140℃时, CO转化率趋近于50%。而引入H2O之后, CO氧化活性被显著抑制, 但当温度达到140℃以上后CO氧化活性略高于无水条件, 转化率趋近于60%。
以Pd (NO3) 2的HBr溶液为前驱体所制备的催化剂BrC, 在无水条件下CO氧化活性高于催化剂C。并且气氛中H2O的进入, 进一步提高了催化剂的CO氧化性能, 在140℃附近趋近于完全转化。
2.1.3 氧化态催化剂的CO氧化原位红外表征
选取C, ClC和BrC 3个催化剂进行了CO吸附的原位红外漫反射表征, 结果如图3~5所示。
对于CO的吸附, 2170~2180 cm-1波数的振动峰归属为Pd2+物种上的CO线式吸附 (Pd2+-CO) , 2090 cm-1波数的振动峰归属为Pd0物种上的CO线式吸附 (Pd0-CO) , 1940~1950 cm-1波数的振动峰归属于Pd+物种上的CO桥式吸附 (Pd2+-CO) , 1860~1870 cm-1波数的振动峰归属于Pd三原子协同吸附CO (Pd3+-CO) , 代表气相CO的振动峰出现在2170及2120 cm-1波数。
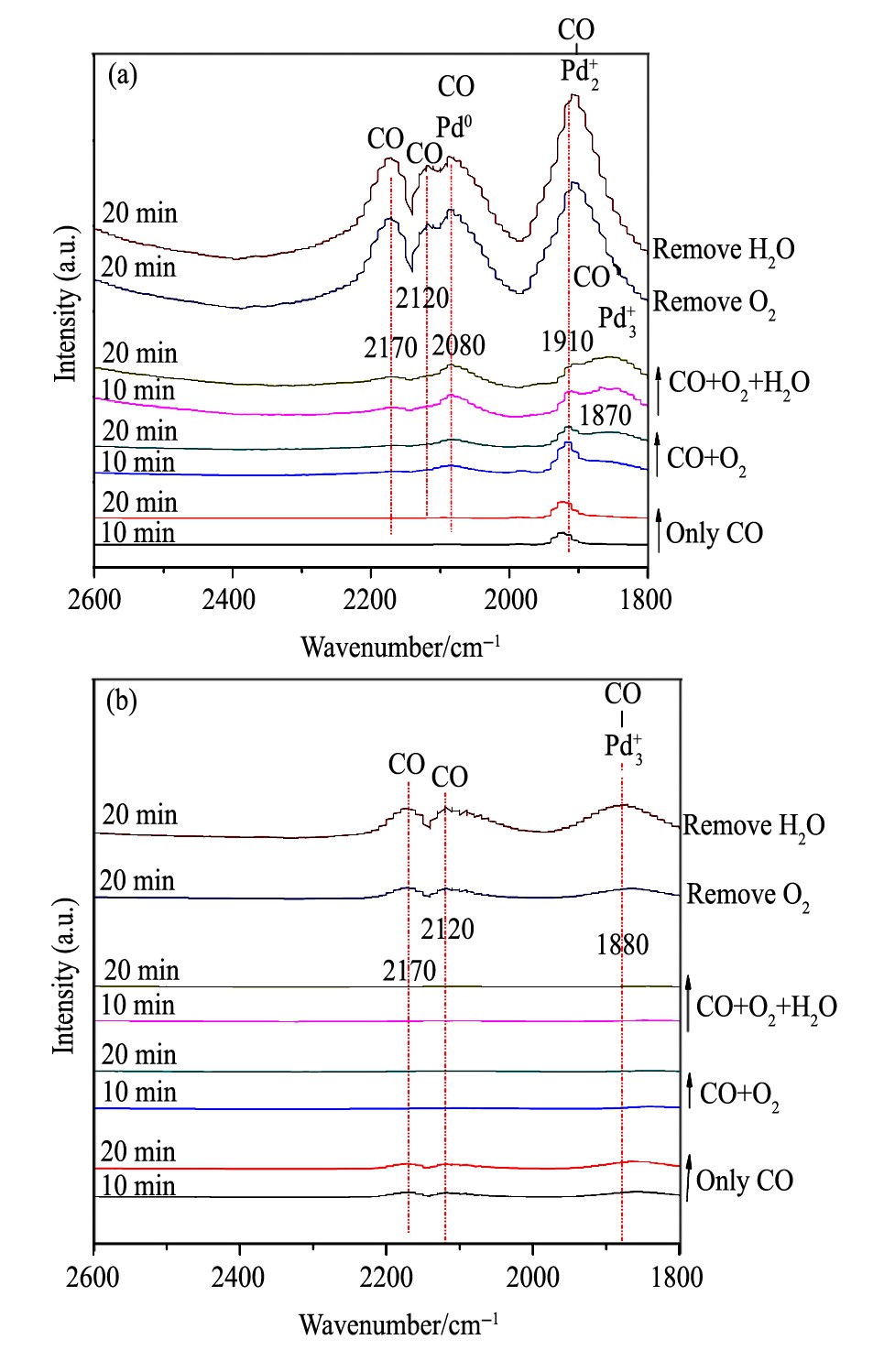
图3 30和90℃温度下催化剂C上CO吸附红外谱图Fig.3 In situ DRIFTS spectra of CO adsorption on C catalysts at 30℃ (a) and 90℃ (b)
图3 (a) 是C催化剂在反应温度为30℃时的CO吸附红外谱图。从图3中可以看出: (1) 通入CO后, 观测到了Pd2+-CO吸附峰。说明当CO引入后, 可促进部分Pd O还原, 形成了Pd+离子。 (2) 当CO+O2共进料时, 随着反应时间的延长, Pd2+-CO的吸附峰逐渐减弱, 同时还检测到较弱的Pd0-CO, 且Pd0-CO的峰强度有增大的趋势。这说明在O2的引入可促进催化剂表面CO的氧化, 同时在此过程中也使得部分Pd物种被进一步还原为金属态Pd。 (3) CO+O2+H2O共进料时, Pd2+-CO的吸附峰快速减弱, 同时Pd0-CO的吸收峰增大, 且出现了Pd3+-CO的吸收峰。这说明H2O的引入有可能促进了表面Pd物种的还原, 并改变了Pd+离子与CO的吸附状态。 (4) 气氛中移除O2后, CO的吸附峰显著增强;进一步移除H2O后, 其CO吸收峰的变化并不明显。以上结果说明30℃时, 在有O2的条件下, H2O对Pd催化剂活性存在一定的影响, 在O2移除后, H2O对Pd催化剂的活性无显著影响。
当反应温度升高至90℃时CO吸附的红外谱图列于图3 (b) 。从图3中可以看出: (1) 通入CO后, 观测到了Pd3+-CO的吸附和气相CO吸收峰, 并且随着吸附时间的延长, 峰强度有所增强。 (2) CO+O2和CO+O2+H2O共进料时, 几乎观测不到CO的吸附峰, 这可能是由于温度过高, 吸附的CO被氧化所致。 (3) 但气氛中O2被移除后, 可观测到Pd3+-CO的吸收峰和气相CO吸收峰。进一步移除H2O后, Pd3+-CO的吸收峰和气相CO吸收峰显著增强。这说明在90℃时, H2O的引入可促进催化剂表面CO的氧化, 这与活性评价结果一致。
对于ClC催化剂, CO在30和90℃的红外吸附分别列于图4 (a) 和 (b) 中。当反应温度为30℃时, (1) 通入CO后, 可以检测到弱的Pd2+-CO吸收峰和气相CO吸收峰, 随着吸附时间的延长该峰逐渐增强。 (2) CO+O2共进料时, 观测到Pd2+-CO的吸收峰转变为Pd3+-CO吸收峰, 且各峰强度均有所降低, 这表明O2的引入促进了CO的氧化, 降低了表面CO吸附的浓度, 使得Pd+对CO的吸附由双原子吸附变为了三原子协同吸附。 (3) 进一步引入H2O, 各CO吸收峰均显著增强, 说明H2O的引入对CO的氧化起到了一定的抑制作用, 这与活性评价相一致。 (4) 当气氛中的O2被移除后, CO吸收峰的强度得到加强且Pd3+-CO的吸收峰转变为Pd2+-CO吸收峰。进一步移除H2O后, CO的吸收峰基本保持不变。这表明O2移除之后Pd表面吸附的CO浓度增多, 且H2O在无氧环境下对CO吸附的影响不大。
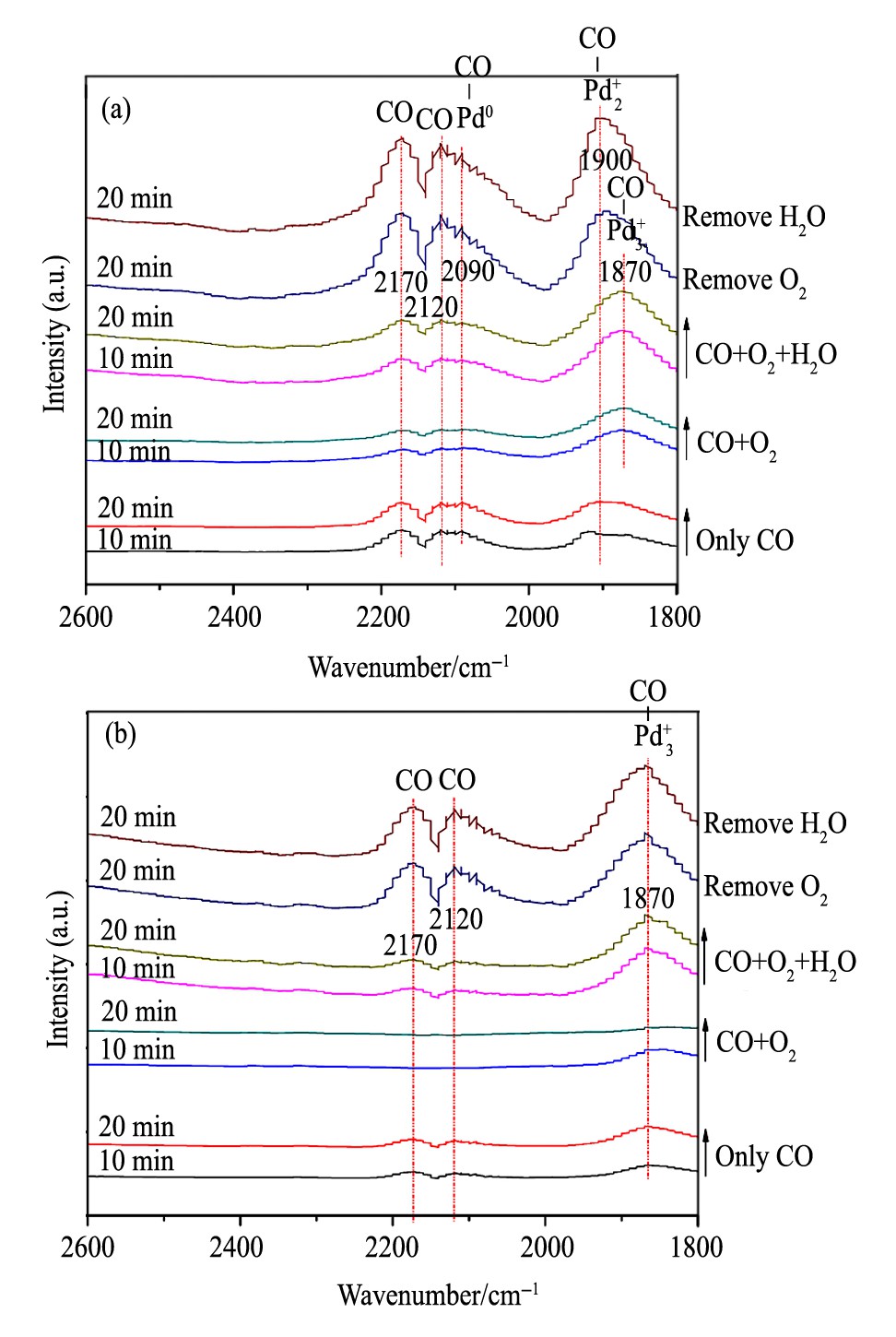
图4 30和90℃温度下催化剂Cl C的CO吸附红外谱图Fig.4 In situ DRIFTS spectra of CO adsorption on Cl C cata-lysts at 30℃ (a) and 90℃ (b)
升高反应温度至90℃时, ClC催化剂上CO吸附性能的变化规律与30℃所得结果类似, 由于高温下更有利于CO的氧化, 在CO与O2共进料时, 几乎观测不到CO的吸附峰。
对于BrC催化剂, CO在30和90℃的红外吸附分别列于图5 (a) 和 (b) 中。当反应温度为30℃时: (1) 通入CO:可以检测到Pd3+-CO吸收峰和气相CO吸收峰。与C催化剂和ClC催化剂相比, CO吸收峰显著增强, 但Pd+离子上吸附的CO主要以Pd3+-CO的形式存在。 (2) 在通入O2后, 各CO吸收峰均有所减弱。进一步引入水后, 各CO吸收峰的减弱并不明显。 (3) 当气氛中O2被移除后, CO吸附峰显著加强, 说明O2的存在可显著促进CO的氧化。进一步移除H2O后, CO吸附峰的基本保持不变。
反应温度为90℃时: (1) 通入CO:可以检测到弱的Pd3+-CO吸收峰和气相CO吸收峰。 (2) CO+O2共进料时, CO吸收峰虽有降低, 但幅度不大。进一步通入H2O后, CO吸收峰有着明显减弱, 说明H2O的通入对CO氧化有着促进作用, 与活性结果一致。 (3) O2被移除后, CO吸收峰的强度有着明显增加, 进一步移除H2O后, CO的吸附强度基本不变, 与之前结果一致。
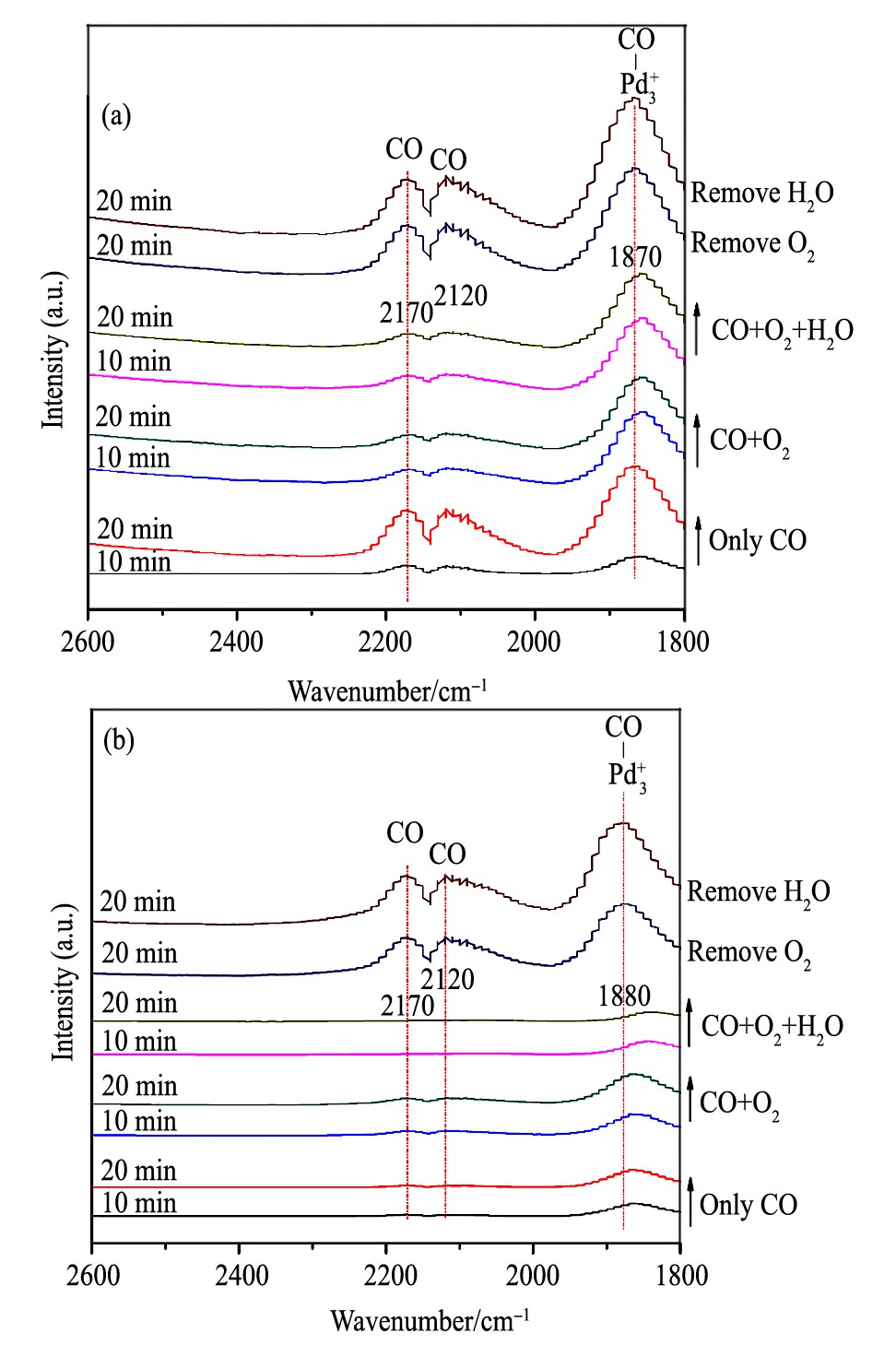
图5 30和90℃温度下催化剂Br C的CO吸附红外谱图Fig.5 In situ DRIFTS spectra of CO adsorption on Br C cata-lysts at 30℃ (a) and 90℃ (b)
2.2 还原态Pd催化剂上CO催化氧化
2.2.1 还原态催化剂的XRD表征
对3种催化剂进行了XRD表征, 结果如图6所示。
CH, ClCH和BrCH的XRD谱图列于图6所示。从图6中看出, 3种样品的XRD图均显现出Al2O3特征峰, 而并没有出现Pd物种特征峰, 这表明Pd物种在催化剂表面均以高分散的形式存在。
2.2.2 还原态催化剂的活性评价
经300℃H2还原3 h后所得催化剂CH, ClCH和BrCH的CO催化氧化的活性评价结果列于图7。
与图2相比, H2还原处理后, 催化剂上CO氧化活性均有显著的提高, 但提高的幅度取决于Pd物种的前驱体形式。
CH催化剂表现出最高的CO氧化活性, 在15℃时CO的转化率约为20%, 最低全转化温度降低至40℃。当Cl离子引入后, 明显抑制了CO氧化, ClCH催化剂上CO转化率在80℃时达到90%, 继续升高温度, CO转化率增加缓慢。对于Br离子的引入, BrCH催化剂在低温区的活性高于ClCH催化剂, CO最低全传温度为100℃。
当反应气氛中引入H2O后, 显著促进了CH催化剂上CO催化氧化的活性, CO最低全转化温度降低至30℃。但对ClCH催化剂, H2O的引入则对CO氧化性能的几乎无影响。而对BrCH催化剂而言, H2O的引入稍许抑制了CO氧化性能。上述结果表明, 对于还原态Pd催化剂, 制备过程中卤素的引入, 可显著影响催化剂的CO氧化活性以及H2O在CO氧化反应中的作用。
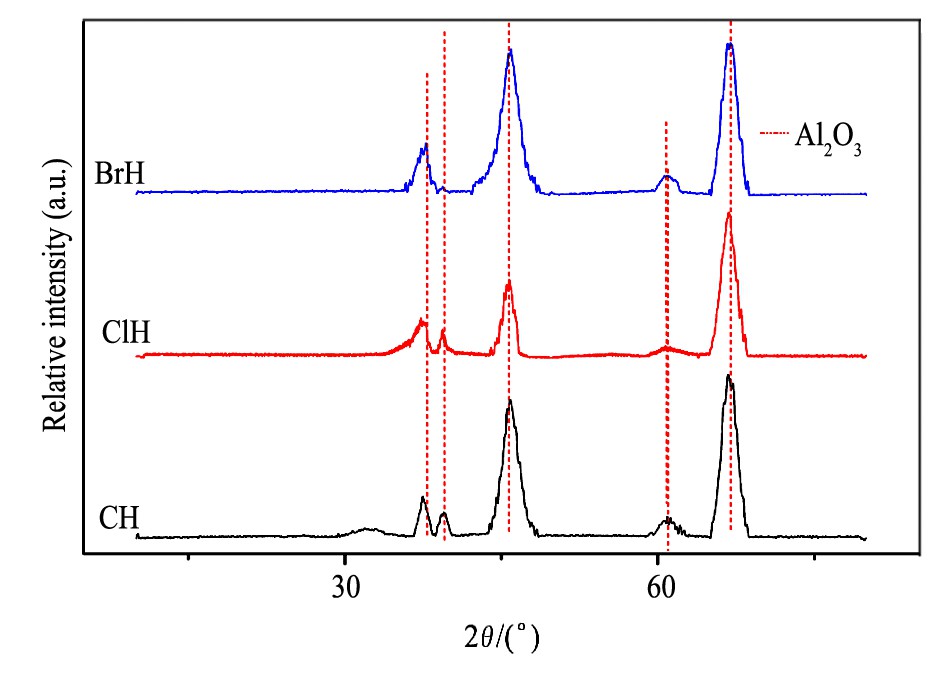
图6 含不同卤素的还原态样品的XRD图Fig.6 XRD patterns of reduced samples containing different halogen
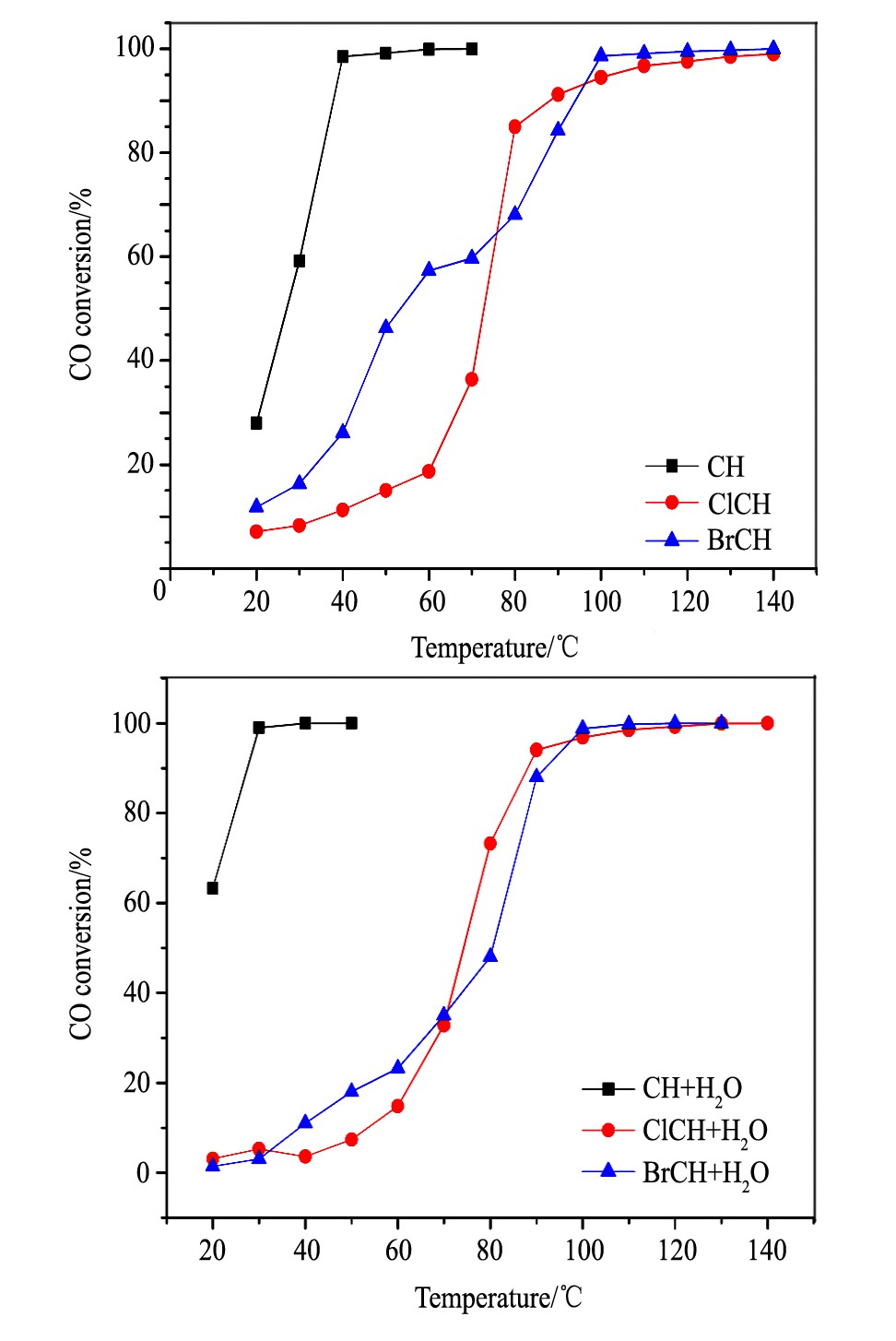
图7 含不同卤素的还原态催化剂的CO氧化活性and after (b) water Fig.7 CO oxidation activity of reduced samples containing dif-ferent halogen before (a) vapor accessing
2.2.3 还原态催化剂的CO吸附原位红外表征
对CH, ClCH和BrCH分别进行了CO吸附原位红外表征。在实验之前先通入H2气体, 升温至200℃对催化剂进行预处理, 自然冷却至30℃。其余步骤与前文一致。结果如图8~10所示。
对于CH催化剂 (图8) , 当反应温度为30℃时: (1) 通入CO后, 观测到了Pd2+-CO吸收峰和宽化的Pd0-CO吸收峰, 说明催化剂虽经H2预处理后, 仍有低价态Pd物种的存在。并且Pd2+-CO吸收峰随吸附时间的延长而增强。 (2) CO+O2共进料时, Pd2+-CO吸收峰的强度显著降低, 同时Pd0-CO吸收峰几乎观测不到。进一步引入H2O后, Pd2+-CO吸收峰的强度继续降低。 (3) 但反应气氛中移除O2后, Pd2+-CO吸收峰显著增强。反应气氛中继续移除H2O后, CO吸附强度进一步升高。说明在此温度下, CH催化剂上的CO氧化反应中O2的作用较为显著, 而H2O也可对CO氧化反应起到促进作用。
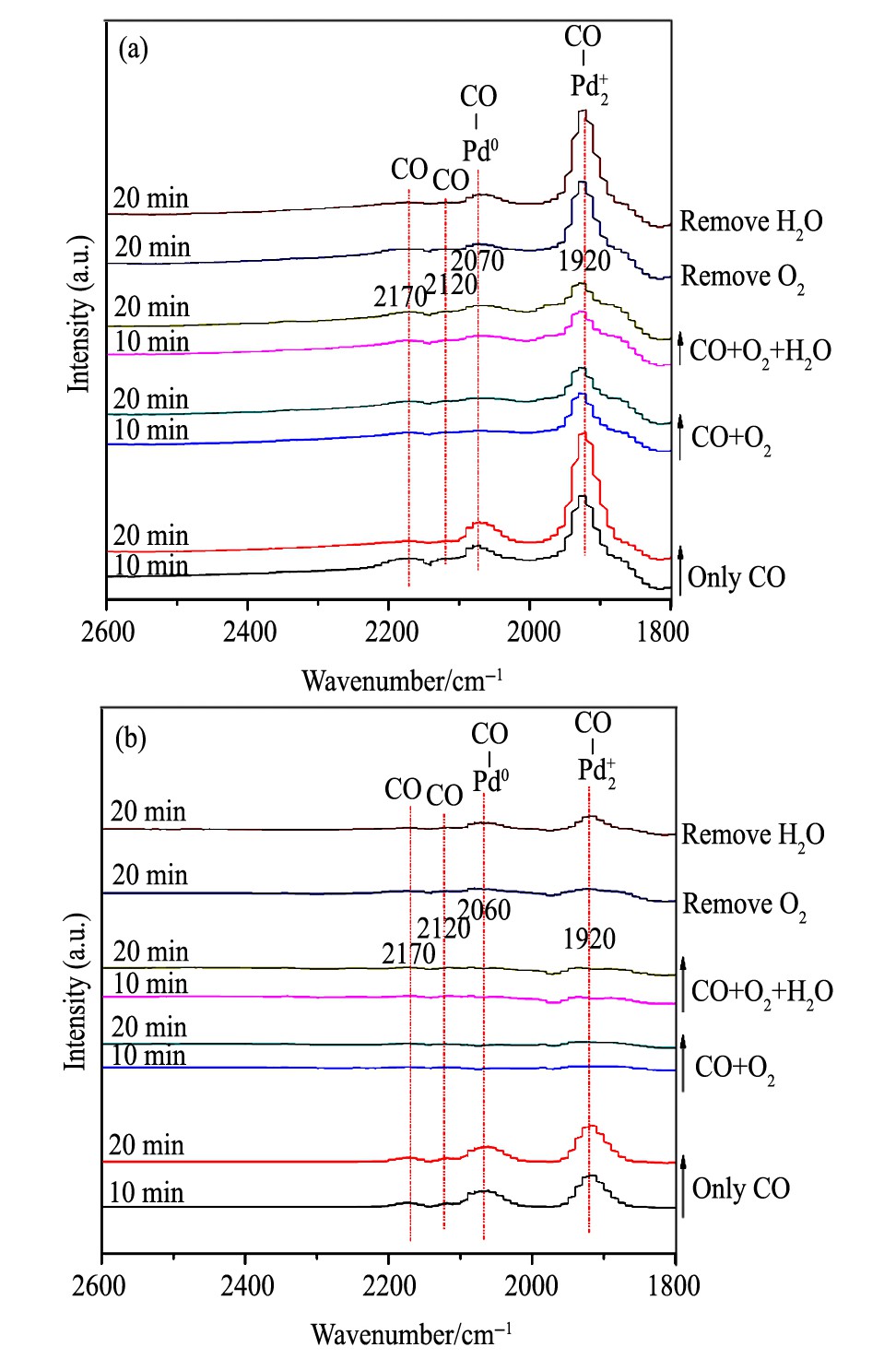
图8 30和90℃温度下CH催化剂的CO吸附红外谱图Fig.8In situ DRIFTS spectra of CO adsorption on CH cata-lysts at 30℃ (a) and 90℃ (b)
当反应温度为90℃时: (1) 随着CO吸附的进行, Pd0-CO吸收峰更为显著, 同时也观测到了Pd2+-CO吸收峰。 (2) 当CO+O2共进料时, Pd0-CO和Pd2+-CO的吸收峰基本消失, 引入H2O后, 红外光谱几乎无变化。结合活性评价结果可认为, 是由此温度下催化剂具有高的CO氧化活性所致。 (3) 当反应气中的O2被移除后, CO的吸收峰几乎无变化, 处了宽化的Pd2+-CO峰强度略有增加。继续移除气氛中的H2O后, Pd2+-CO峰强度有所提高, 同时观测到Pd0-CO的吸收峰。
对于ClCH催化剂 (图9) , 在反应温度为30℃时: (1) 随着CO吸附的进行, 可观测到Pd2+-CO的吸收峰、Pd0-CO吸收峰和气相CO吸收峰。 (2) 当CO+O2共进料时, Pd2+-CO吸收峰显著减弱, 其余CO吸收峰基本消失。进一步引入H2O后, 各CO吸收峰几乎保持不变, 说明在此温度下, H2O对反应活性的影响不显著。 (3) 气氛中的O2被移除后, Pd2+-CO吸收峰显著增大, 并且该吸收峰随着H2O的移除, 其强度进一步增强, 与之前的结果一致。
当反应温度升至90℃时, (1) CO吸附的峰型与30℃时相似。 (2) 通入O2后CO吸收峰迅速减弱, 进一步引入H2O后, 其CO吸附性能基本保持不变。 (3) O2移除后CO吸收峰迅速增大, 进一步移除H2O后, 峰强度变化不明显。这说明与CH催化剂对比, H2O对CO氧化的促进作用明显减弱。对于BrCH催化剂 (图10) , 在反应温度为30℃时: (1) 随着CO吸附的进行, 观测到了显著的Pd2+-CO吸收峰和微弱的Pd0-CO吸收峰。 (2) 在气氛引入O2后, Pd0-CO吸收峰消失同时Pd2+-CO吸收峰的强度减小较少。通水后CO吸附峰强度基本无变化, 说明该温度下H2O对催化反应活性的影响很小, 这与活性结果一致。 (3) 气氛中的O2被移除后, Pd2+-CO吸收峰显著升高。随着气氛中的H2O被移除后, CO吸附峰的强度基本保持不变, 与之前的研究一致。
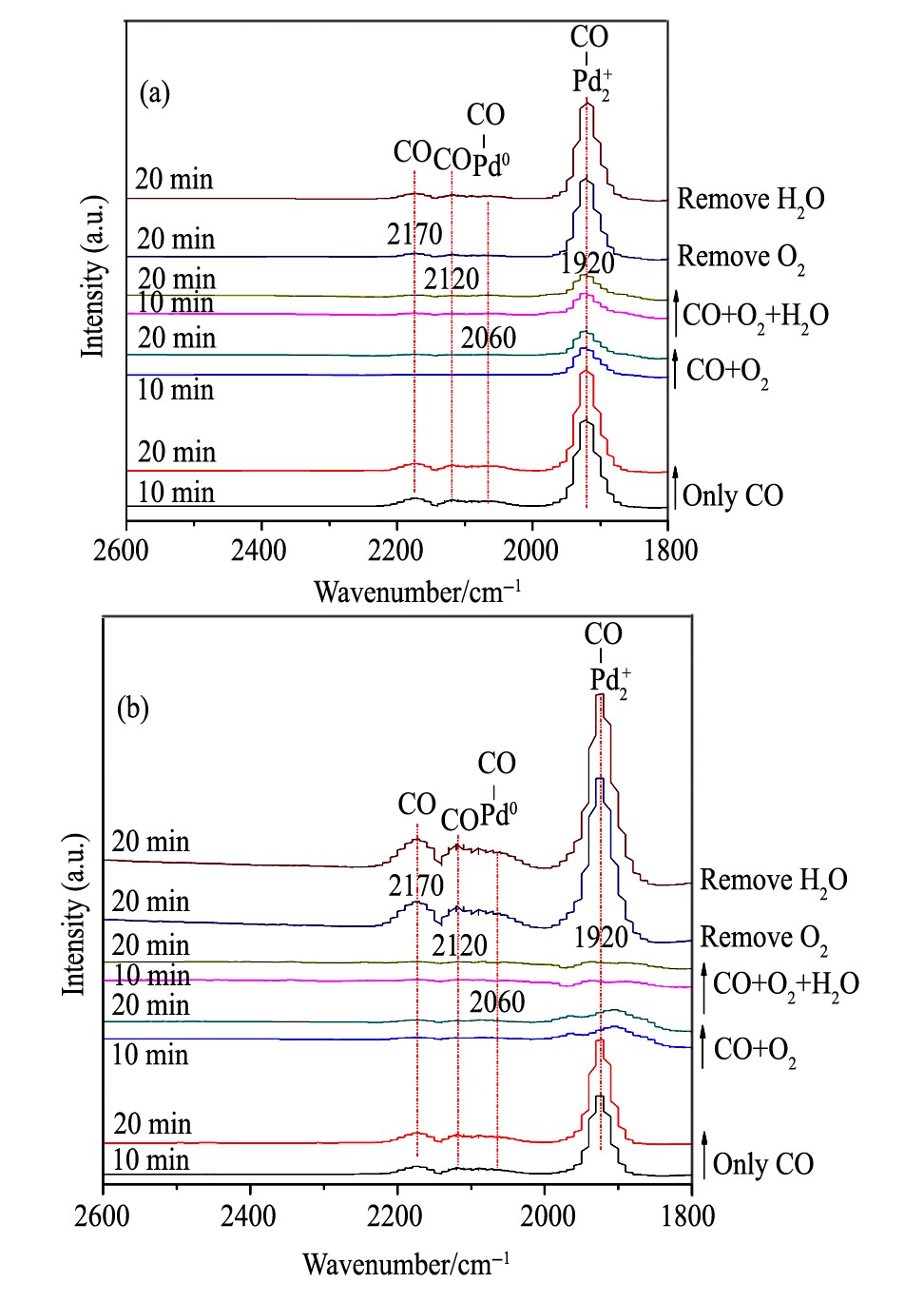
图9 30和90℃温度下Cl CH催化剂的CO吸附红外谱图Fig.9 In situ DRIFTS spectra of CO adsorption on Cl CH cata-lysts at 30℃ (a) and 90℃ (b)
当反应温度为90℃时: (1) 随着CO吸附的进行, 除了与之前类似的峰型以外, 在1980 cm-1的位置上出现了一个新的吸收峰, 该吸收峰归为Pd20-CO吸收峰。这说明在Br-离子引入后, 在金属态Pd表面的CO吸附浓度降低, 出现了桥式吸附CO的状态。 (2) 通入氧气后各CO吸收峰强度显著降低, 通水后CO吸收峰有着一定的增大, 说明H2O的引入对反应起到了一定的抑制作用, 这与活性结果一致。 (3) 气氛中的O2被移除后, 各CO吸收峰显著升高。移除H2O后峰强无明显变化, 与之前研究一致。
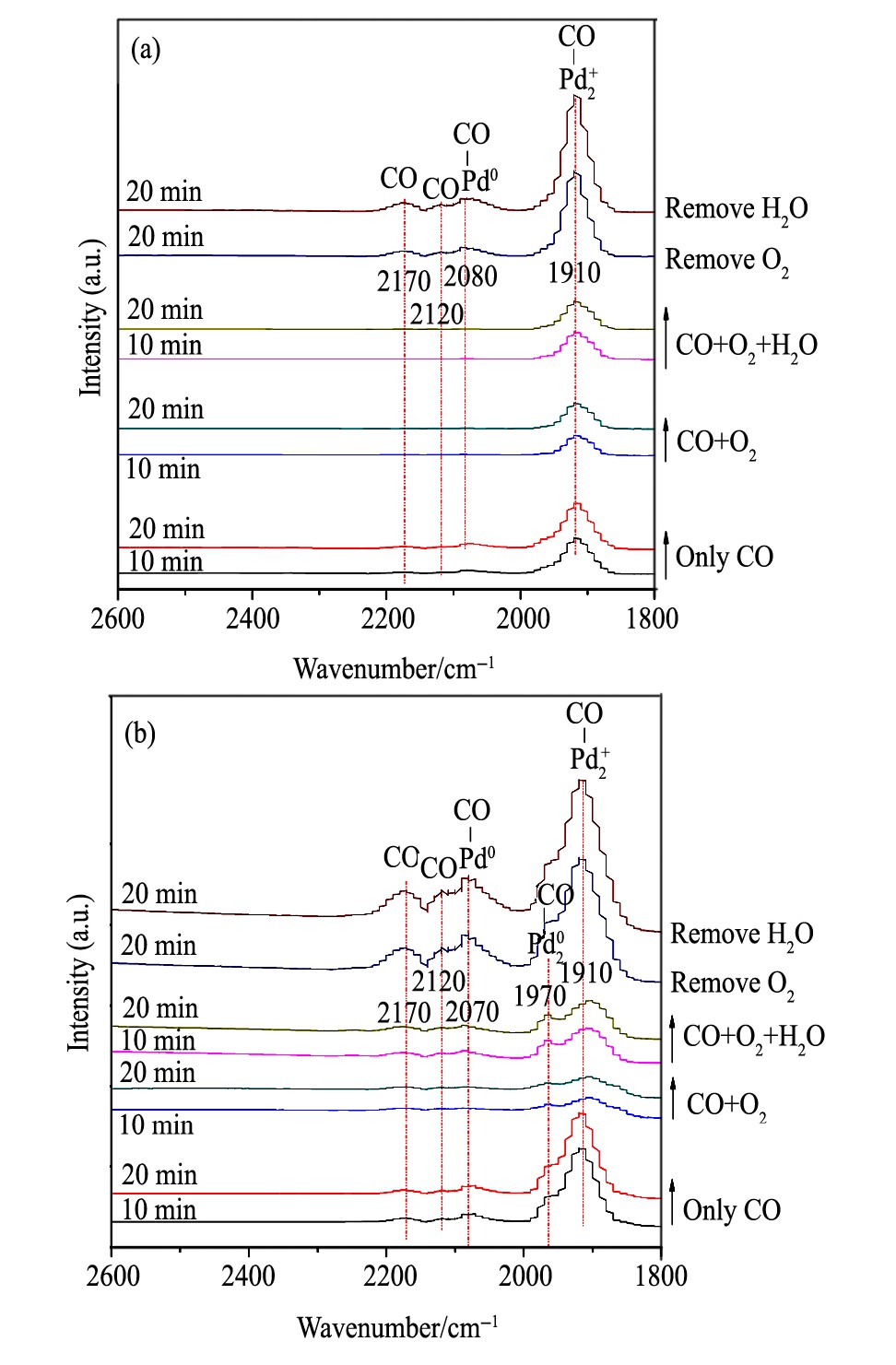
图1 0 30和90℃温度下Br CH催化剂的CO吸附红外谱图Fig.10 In situ DRIFTS spectra of CO adsorption on Br CH cat-alysts at 30℃ (a) and 90℃ (b)
上述研究表明, Pd物种的前驱体对CO氧化反应性能均有较为显著的影响。当Pd以氧化态存在时, 以PdCl2的HCl溶液为前驱体所得ClC催化剂的活性最低, BrC催化剂具有最高的反应性能。当催化剂经H2还原后, 催化剂的活性均有显著的提高, 其中CH表现出最高的CO氧化活性。
原位红外表征结果表明, 当Pd物种以还原态形式存在时, 在CO吸附过程中催化剂表面主要以Pd2+-CO和Pd0-CO的形式存在。气氛中引入H2O后, CH催化剂表面的CO吸附物种显著减少 (图8) , 说明H2O的引入抑制了CO的吸附。此时催化剂表面存在的H2O可以通过-OH与CO反应, 同时还可有利于O2的吸附和活化, 从而显著提高了CO的氧化活性。当Cl和Br引入后, ClCH和BrCH催化剂表面CO吸附物种同样以Pd2+-CO和Pd0-CO的形式存在。但H2O的存在对CO吸附物种的影响不大 (图9和10) , Pd2+-CO和Pd0-CO基本保持不变, 因此H2O的引入对其活性影响不明显, 这也活性评价结果一致。
但对于Pd物种以氧化态形式存在时, H2O对CO的影响则表现的更为复杂。在低温CO吸附过程中, 对于C催化剂而言, 主要检测到了Pd2+-CO物种, ClC催化剂表面除了Pd2+-CO外, 还可检测到Pd0-CO和Pd3+-CO的存在, 而BrC催化剂则以Pd3+-CO物种为主。这说明Cl和Br的引入可促进Pd物种的还原。结合还原态Pd催化剂所得结论, 似乎H2O对ClC和BrC催化剂上CO氧化的促进作用应更为明显。但评价结果表明H2O可促进C和BrC催化剂上CO的氧化, 其中BrC催化剂上的促进作用更为显著。但ClC催化剂上却在室温至140℃的范围内抑制了CO的氧化。
当Pd以氧化态形式存在时, 其表面难以吸附和活化O2[17], 此时CO氧化反应可认为遵循的是M.K.机制, 即表面吸附的CO与表面晶格氧反应, 同时表面Pd物种被还原, 然后O2继续在还原的Pd物种上继续吸附活化, 从而完成反应循环。原位红外表明, 在低温阶段 (30℃) CO+O2和CO+O2+H2O共进料时, 对于C催化剂, 除了Pd2+-CO物种外, 还检测到了Pd0-CO和Pd3+-CO的存在, ClC催化剂表面则以Pd3+-CO物种为主, 而BrC催化剂表面CO的吸附形式则无显著变化, 依然以Pd3+-CO物种的形式存在。但升高反应温度至90℃时, H2O引入显著减少了C和BrC催化剂表面吸附的CO, 而ClC催化剂表面的Pd3+-CO物种却有显著的增加, 说明H2O的引入抑制了该催化剂表面CO的氧化, 这可能与所引入的Cl影响了Pd物种的存在形式和吸附性能。关于这点还需进一步的研究。
3 结论
采用等体积浸渍的方法, 制备了一系列Pd/Al2O3体系的催化剂, 研究了Pd的化学状态、卤素离子以及H2O的引入对CO催化氧化反应性能的影响。得到以下结论:
1.Pd物种的前驱体及存在状态对CO氧化活性有显著的影响。当Pd以氧化态存在时, 以PdCl2的HCl溶液为前驱体所得ClC催化剂的活性最低, BrC催化剂具有最高的反应性能;当催化剂经H2还原后, 催化剂的活性均有显著的提高, 其中CH表现出最高的CO氧化活性。
2.H2O对催化剂上CO氧化性能的影响不仅取决于Pd物种的存在状态, 同时Cl/Br的引入对其也有显著的影响。对于还原态的催化剂, H2O的引入抑制了表面CO的吸附, 并有利于O2的吸附和活化, 从而显著提高了CO的氧化活性。但Cl和Br引入后, H2O的存在对CO吸附几乎无影响, 进而对其活性影响不明显。当Pd以氧化态形式存在时, H2O的引入可促进BrC和C催化剂上CO的催化氧化, 其中BrC催化剂上的促进作用最为明显, 但却抑制了ClC催化剂上的CO氧化。这可能与所引入的Cl影响了Pd物种的存在形式和吸附性能有关。
参考文献
[1] Jernigan G, Somorjai A. Carbon monoxide oxidation over three different oxidation states of copper:metallic copper, copper (I) oxide, and copper (II) oxide―a surface science and kinetic study[J]. Journal of Catalysis, 1994, 147 (2) :567.
[2] Jansson J. Low-temperature CO oxidation over Co3O4/Al2O3[J]. Journal of Catalysis, 2000, 194 (1) :55.
[3] Xie X, Li Y, Liu Z Q, Haruta M, Shen W. Low-tem-perature oxidation of CO catalysed by Co3O4nanorods[J]. Nature, 2009, 458 (9) :747.
[4] Buciuman F C, Patcas F, Hahn T. A spillover approach to oxidation catalysis over copper and manganese mixed oxides[J]. Chemical Engineering and Processing:Process Intensification, 1999, 38 (4) :563.
[5] Güttel R, Paul M, Galeano C, Schüth F. Au, @Zr O2yolk-shell catalysts for CO oxidation:study of particle size effect by expost size control of Au cores[J]. Journal of Catalysis, 2012, 289:100.
[6] Wang R, He H, Wang J, Liu L, Dai H. Shape-regulation:an effective way to control CO oxidation activity over noble metal catalysts[J]. Catalysis Today, 2013, 201 (1) :68.
[7] Bogdanchikova N, Pestryakov A, Tuzovskaya I, Zepeda T A, Farias M H, Tiznado H, Martynyuk O. Effect of redox treatments on activation and deactivation of gold nanospecies supported on mesoporous silica in CO oxidation[J]. Fuel, 2013, 110:40.
[8] Schubert M M, Hackenberg S, van Veen A C, Muhler M, Plzak V, Behm R J. CO oxidation over supported gold catalysts―“Inert”and “Active”support materials and their role for the oxygen supply during reaction[J].Journal of Catalysis, 2001, 197 (1) :113.
[9] Njagi E C, Chen C H, Genuino H, Galindo H, Huang H, Suib S L. Total oxidation of CO at ambient temperature using copper manganese oxide catalysts prepared by a redox method[J]. Applied Catalysis B:Environmental, 2010, 99 (1) :103.
[10] Chen M S, Cal Y, Yan Z, Gath K K, Axnanda S, Goodman D W. Highly active surfaces for CO oxidation on Rh, Pd, and Pt[J]. Surface Science, 2007, 601 (23) :5326.
[11] Li C X, Leong W K, Zhong Z Y. Metallic osmium and ruthenium nanoparticles for CO oxidation[J]. Journal of Organometallic Chemistry, 2009, 694 (15) :2315.
[12] Bergeld J, Kasemo B, Chakarov D V. CO oxidation on Pt (111) promoted by coadsorbed H2O[J]. Surface Science, 2001, 495 (3) :815.
[13] Zhou H P, Wu H S, Shen J, Yin A X, Sun L D, Yan C H. Thermally stable Pt/Ce O2hetero-nanocomposites with high catalytic activity[J]. Journal of the American Chemical Society, 2010, 132 (14) :4998.
[14] Kan H H, Shumbera R B, Weaver J F. Adsorption and abstraction of oxygen atoms on Pd (111) :characterization of the precursor to Pd O formation[J]. Surface Science, 2008, 602 (7) :1337.
[15] Gopinath R, Babu N S, Kumar J V, Lingaiah N, Prasad P S S. Influence of Pd precursor and method ofpreparation on hydrode chlorination activity of alumina supported palladium catalysts[J]. Catalysis Letters, 2008, 120 (3-4) :312.
[16] Shen Y X, Guo Y, Wang L, Wang Y Q, Guo Y L, Gong X Q, Lu G Z. The stability and deactivation of Pd-Cu-Clx/Al2O3catalyst for low temperature CO oxidation:an effect of moisture[J]. Catalysis Science&Technology, 2011, 1 (7) :1202.
[17] Wang H F, Kavanagh Richard, Guo Y L, Guo Y, Lu G Z, Hu P. Structural origin:water deactivates metal oxides to CO oxidation and promotes low-temperature CO oxidation with metals[J]. Angewandte Chemie, 2012, 51 (27) :6657.

