Trans. Nonferrous Met. Soc. China 24(2014) 582-587
On-line detection of Cu (II) in bioleaching system by anodic stripping differential pulse voltammetry
Yan JIN1,2,3, Miao CHEN2, Qing-hui JIN1, Jian-long ZHAO1
1. State Key Laboratory of Transducers Technology, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai 200050, China;
2. CSIRO Process Science and Engineering, Box 312, Clayton South, Victoria 3169, Australia;
3. University of Chinese Academy of Sciences, Beijing 100049, China
Received 5 January 2013; accepted 21 November 2013
Abstract: On-line Cu (II) ion concentration detection in bioleaching system was achieved by anodic stripping differential pulse voltammetry (ASDPV). Good linearity between Cu (II) concentration and oxidation peak current was obtained when Cu (II) existed in 0K media in the concentration range of 1 ��mol/L (64 ��g/L) to 1 mmol/L (64 mg/L). Moreover, when 0.2 mol/L KCl was added into this media, the linear detection range could be extended from 1 mmol/L to 100 mmol/L (6.4 g/L). The reduction of Cu (II) to metallic copper was shown to proceed as two successive single-electron transfer reactions involving an intermediate chemical step where the cuprous ion (Cu+) was complexed by chloride to form the dichlorocuprous anion (CuCl-). In addition, interference effect was also investigated when Fe3+ existed in the media, which was the common situation in the copper bioleaching system. The results showed no interference effect once the concentration of Fe3+ was less than 100 mmol/L (5.6 g/L).
Key words: anodic stripping differential pulse voltammetry (ASDPV); peak current; Cu (II); Fe3+ interference
1 Introduction
Bioleaching is an emerging technology with significant potentials to add value to the mining industries so as to deliver attractive environmental and social benefits [1]. Low-grade ores, small and complicated ore bodies, waste ores and ores that are hard to be processed by traditional method can be recovered by bioleaching. Compared with traditional strong acid chemical leaching method, bioleaching has many advantages, such as economic, energy-efficient, pollution-free and low capital requirements, particularly when the recoverable metals are present at relatively low levels [2,3].
The concentration of Cu (II) is a vital factor that reflects the copper leaching rate, recovery efficiency and influences the next processions, such as solvent extraction and electrowinning [3,4]. There is a strong requirement and application for on-line Cu (II) ion concentration detection. Different methods have been employed for metal analysis in bioleaching, such as atomic absorption spectrometry (AAS) [5,6], inductively coupled plasma emission spectrometry (ICP-AES) [6], energy dispersive X-ray fluorescence (EDXRF) [7,8] and total reflection X-ray fluorescence (TXRF) [9]. These methods are of high sensitivity. However, these methods are limited in their use for in-situ and on-line monitoring because of their size, cost and analysis time.
Copper ion-selective electrodes give a linear, Nernstian response to Cu2+ activities, and could be used for on-line monitoring but they are subject to a number of interferences. Their precision and accuracy can be reduced by the presence of interfering ions, temperature variations and electrode surface reactions [10,11]. Commercial copper ion-selective electrode failed to work in the presence of iron at low pH value, thus they are hard to be used in the on-line bioleaching system.
Electrochemical techniques, such as stripping analysis, have been reported for the heavy metal analysis because they satisfy almost all requirements for in-situ measurement of heavy metal ions [12-15], and are capable of detecting more than one electro-active species at the same time. Anodic stripping voltammetry (ASV) and cathodic stripping voltammetry (CSV) are the two typical representative stripping analysis methods. By ASV, the analyte of interest is electroplated on a working electrode during a deposition step, and then oxidized from the electrode during a subsequent stripping step, where the oxidation current is measured. The oxidation of species is registered as a peak in the current signal associated with the potential at which the species is known to be oxidized [16]. By CSV, heavy metal ions in the sample solution can be identified and quantified by sweeping a potential that reduces specific metal ions at a specific potential and the current generated at each reduction potential indicates metal ion species and concentration on the order of minutes [17,18].
A number of different pulse techniques are available for stripping voltammetry. Differential pulse voltammetry (DPV) is selected for our experiments, where the working electrode current is measured prior to and at the end of a pulse superimposed on a staircase potential ramp, with the difference signal recorded. DPV is generally accepted as being more sensitive than conventional cyclic voltammetry. It can discriminate against the large capacitive currents associated with the charging and discharging of the electrical double layer on the electrode surface where the currents are recorded as the electrode potential is rapidly changed during a voltammetric sweep [19].
In this study, the concentration of Cu (II) is measured by DPV, in an attempt to find an on-line measurement method suitable for the determination of Cu (II) ion concentrations in the bioleaching system.
2 Experimental
2.1 Apparatus and materials
Differential pulse stripping voltammetric analyses were performed with a CHI scanning electrochemical microscope (CHI instruments, USA). A standard three-electrode configuration was used for the electrochemical measurements, a glassy carbon electrode (electrode surface area was 0.07 cm2), a Pt plate counter electrode and an Ag/AgCl (3 mol/L KCl) reference electrode. All potentials were reported versus the Ag/AgCl (3 mol/L KCl) reference electrode at room temperature. The measurement parameters for ASDPV were chosen as follows: deposition time 60 s with -500 mV, scan rate 50 mV/s, sampling width 17 ms, pulse amplitude 50 mV, pulse period 200 ms, quiet time 10 s and sensitivity 10-5 A/V.
In the experiments reported, the background electrolyte was 0K media, which contained (NH4)2SO4 (3.0 g/L), MgSO4��7H2O (0.5 g/L), K2HPO4 (0.5 g/L), KCl (0.1 g/L) and Ca(NO3)2 (0.01 g/L). Cu2+ and Fe3+ stock solutions were prepared by dissolving cupric sulfate and ferric sulfate into 0K media, respectively. Both pH values were adjusted to 1.8. All chemicals were used as supplied with no further treatment and were of analytical grade. De-ionized water (resistance over 18 M����cm) from a Millipore-Q water purification system was used throughout the experiment.
2.2 Measurement procedure
In this investigation, the cupric and ferric concentrations were both varied over the range of 1 ��mol/L to 100 mmol/L. This range was chosen from the regular cupric and ferric concentration in bioleaching system. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) were applied to achieving cupric concentration detection.
Prior to performing experiments with cupric ions, the glassy carbon electrodes were characterized in the background electrolyte (0K media) to establish the potential limits over which they could be operated without hydrolysis of water.
3 Results and discussion
3.1 CV of 0K media and Cu (II)
Figure 1 shows CV waveforms obtained in the background 0K media (pH 1.8) and 0K media containing 2 mmol/L Cu2+ recorded over the potential ranging from 800 mV to -800 mV and back to 800 mV. It is shown that the glassy carbon electrode could be safely operated at anodic potentials up to 800 mV without oxygen evolution occurring. However, the presence of large currents at potentials more negative than approximately -500 mV suggests that this potential sets the cathodic limit before hydrogen evolution commences, as shown in the inserted figure in Fig. 1.
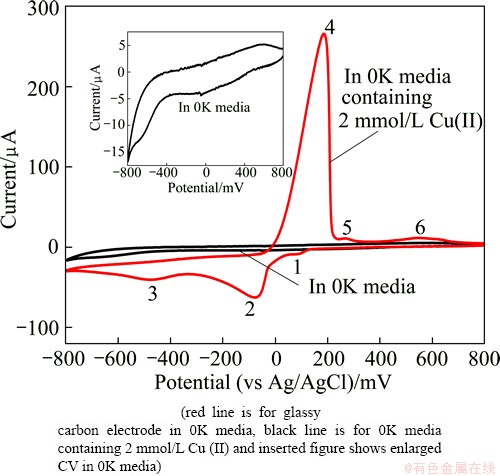
Fig. 1 Cyclic voltammetry waveform
Six peaks appear in the CV waveforms for 0K media containing 2 mmol/L Cu2+. Peak 2 is the reduction of Cu2+ to metallic copper and peak 4 represents the oxidation of metallic copper to Cu2+. As can be seen, the oxidation current (peak 4) is much larger than the reduction current (peak 2), so ASV was selected as the DPV operating method, an anodic stripping differential pulse voltammetry (ASDPV), which is used to characterize the cupric response in the following experiments. Peak 3 can be ascribed to the hydrogen evolution process as its occurring potential is quite close to -500 mV. Peak 6 is considered to be the background effect since CV waveforms for other Cu2+ concentrations (1 mmol/L, 5 mmol/L) also show the same peak. Peak 1 and peak 5 can be caused by the Cl- effect. About 1 mmol/L Cl- exists in 0K media, and more details about Cl- effect will be discussed in section 3.3.
3.2 DPV experiments in 0K media containing Cu (II)
Compared with CV, DPV has better selectivity. The potential ranging from -500 mV to 800 mV was chosen for ASDPV to characterize the cupric response. For each experiment, the working electrode was cleaned by holding the potential at 800 mV for 60 s.
When the electrodes were immersed in 0K media containing Cu (II) and the potential was set at -500 mV for 60 s, Cu (II) was reduced to Cu (0). As can be seen from Fig. 2, an anodic peak at about 180 mV vs Ag/AgCl electrode, corresponding to the oxidation of reduced Cu (0) to Cu (II) is observed. The oxidation peak current increases respectively with increasing concentration of cupric ions from 1 ��mol/L to 5 mmol/L. Linear calibration plot is obtained with correlation coefficient of 0.987 in the concentration range of 1 ��mol/L to 1 mmol/L, as shown in the inset of Fig. 2. So the linearity range for Cu (II) was from 64 ��g/L to 64 mg/L.
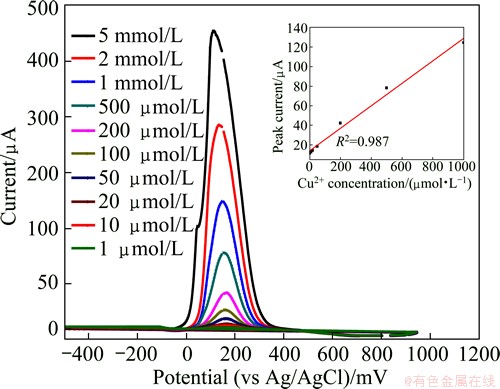
Fig. 2 ASDPV for Cu (II) in range of 1 ��mol/L to 5 mmol/L (the inset is its calibration curve)
3.3 DPV experiments of Cu (II) in 0K media containing Cl-
To understand the influence of Cl-, a series of 0K media containing 0.2 mol/L KCl with different concentrations of cupric ion were tested by cyclic DPV, and the results are shown in Fig. 3(a). The voltammograms show four distinct peaks: two reduction peaks during the cathodic scan (peak 1, peak 2) and two oxidation peaks during the reverse scan (peak 3, peak 4). The presence of four peaks implies the occurrence of two distinct redox processes. It has been reported that the current peaks correspond to the following reactions: the reduction of Cu2+ to Cu+ in solution (peak 1); the reduction of Cu+ to metallic copper at the glassy carbon electrode (peak 2); the oxidation of metallic copper from the glassy carbon electrode (peak 3); and finally the oxidation of Cu+ to Cu2+ in solution (peak 4) [19-22]. During the investigation, a copper film can be observed to cover the surface of glassy carbon electrode during the formation of peak 2 which is subsequently removed during the formation of peak 3, confirming that these two current peaks represent the plating and stripping of copper.
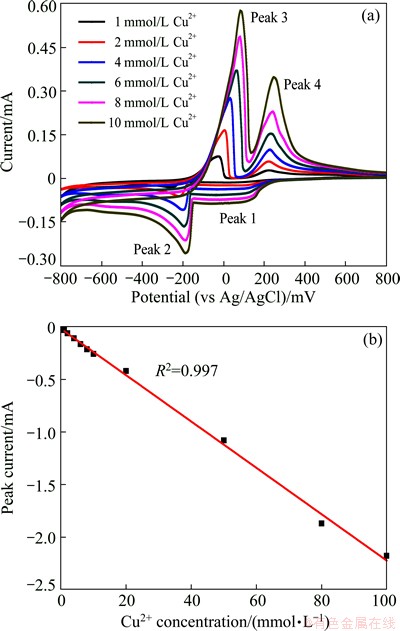
Fig. 3 Cyclic DPV waveform of 0K media containing Cu (II) in the presence of 0.2 mol/L KCl (a) and linear curve (b)
Figure 3(a) illustrates some general trends that are observed over the concentration range studied by cyclic DPV. All four peaks exhibit an increase in magnitude with increasing Cu2+ ion concentration. Moreover, the potential of peak 2 remains constant while the potentials at which peak 3 occurs shifts toward more positive values with concentrations increasing. Peak 2 is used for cupric ion concentration detection.
The linear dependence of peak 2 current (Ip) gives the regression equation of Ip=-0.915-0.025 [Cu (II)], where Ip is in ��A, and [Cu (II)] is in ��mol/L with a correlation coefficient of 0.998, and ranges from 1 mmol/L to 10 mmol/L. It shows that the addition of 0.2 mol/L KCl could increase the linear detection range. Catholic stripping DPV (CSDPV) was used to investigate peak 2 current for 20 , 50 and 100 mmol/L Cu (II) in 0K media containing 0.2 mol/L KCl, and the results are shown in Fig. 3(b). Good linear characteristic is obtained with a correlation coefficient of 0.997. Therefore, the addition of Cl- into this system can increase the Cu (II) detection range to 100 mmol/L (6.4 g/L).
The characteristic 4-peak voltammograms displayed in Fig. 3(a) have been reported [20-23]. In general, the voltammograms represent two separate single electron transfer reactions. The reduction may proceed as two successive single-electron reactions where Cu+ is as an intermediary:
Cu2++e��Cu+, ~ -47 mV (vs Ag/AgCl, 3 mol/L KCl) (1)
Cu++e��Cu, ~ -311 mV (vs Ag/AgCl, 3 mol/L KCl) (2)
The presence of 4-peak suggests a two-stage process, but the potentials at which the observed current peaks occur do not agree with those given by reactions (1) and (2), which was obtained from the Eh-pH diagram for Cu/Cu+/Cu2+ in water [24]. It could be concluded that the redox of Cu2+ must be affected by the chloride ion.
The cuprous ion (Cu+) is not stable in aqueous media and is easily oxidized [25]. The presence of chloride ions, stabilizes it in a highly soluble dichlorocuprous anion 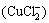 [19,20,26]:
[19,20,26]:
Cu++2Cl-�� (3)
(3)
Thus, the second reduction reaction given by reaction (2) is unlikely to occur in electrolytes with a high chloride concentration and therefore the reduction of Cu2+ more likely proceeds as follows:
Cu2++e��Cu+ (Peak 1) (4)
Cu++2Cl-�� (5)
+e��Cu+2Cl- (Peak 2) (6)
In general, in 0K media with 0.2 mol/L KCl, the reduction of Cu2+ to metallic copper proceeds as two successive single-electron reactions are separated by an intermediate chemical step where the cuprous ion (Cu+) is converted to the dichlorocuprous anion .
3.4 DPV experiments of Cu (II) and Fe (III) in 0K media
Figure 4 shows a group of CV curves of Cu (II) and Fe (III) in 0K media. When the concentration of Fe (III) is less than 1 mmol/L, the interference of Fe (III) can be neglected as the redox peak currents for Cu (II) and Fe (III) were separated. Peaks 1 and 2 are considered to be the redox process for Cu (II) and peaks 3 and 4 for Fe (III). However, when the concentration of Fe (III) is higher than 2 mmol/L, the redox peaks for Cu (II) and Fe (III) overlappes. DPV details were investigated for the Fe (III) concentration larger than 1 mmol/L.
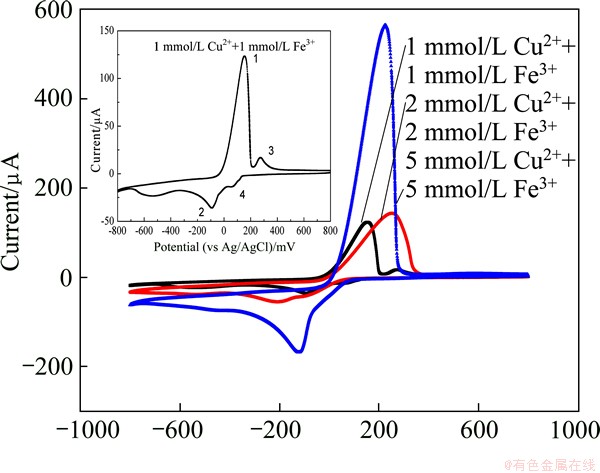
Fig. 4 CV curves of Cu (II) and Fe (III) in 0K media
DPV waveforms of 9K media containing 1 mmol/L Fe (III) with different Cu (II) concentrations are shown in Fig. 5(a). As can be seen, Cu (II) concentrations can be detected when interference Fe (III) is 1 mmol/L. The linear dependence of oxidation peak current on the cupric ions concentration gives the regression equation of Ip=6.633+0.463 [Cu (II)] with a correlation coefficient of 0.971. The detection limit value for cupric ion is determined to be 10-7 mol/L, and the sensitivity will become better when the cupric ion concentration is higher than 10 ��mol/L.
To detect the interference of higher concentration of Fe (III), same concentrations of Cu (II) and Fe (III) have been investigated by DPV, as shown in Fig. 6(a). Anodic peaks at around 80 mV are observed corresponding to the oxidation of Cu (0) to Cu (II). Linear calibration plot (Fig. 6(b)) is Ip=0.349+0.0757 [Cu (II)], where Ip is in mA, and [Cu (II)] is in mmol/L with a correlation coefficients of 0.985. Cu (II) concentration can be detected once Fe3+ ion concentration is lower than 100 mmol/L, that is 5.6 g/L, which is suitable for the bioleaching system.
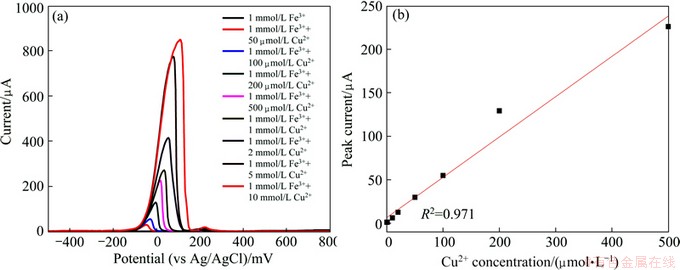
Fig. 5 DPV waveforms of Cu (II) in 0K media with 1 mmol/L Fe (III) (a) and calibration curve (b)
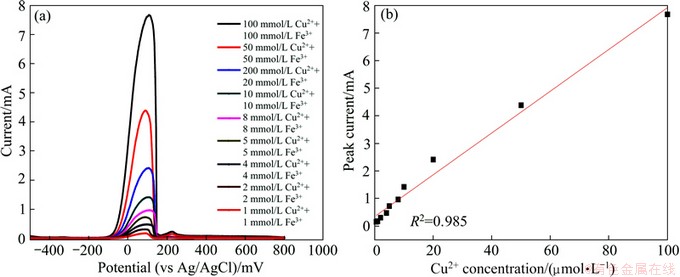
Fig. 6 DPV waveforms for Cu (II) and Fe (III) in 0K media (a) and calibration curve (b)
4 Conclusions
Good linearity was obtained when cupric ions existed in 0K media with the range of 1 ��mol/L to 1 mmol/L in the proposed DPV test. Experimental results showed that in 0K media containing 0.2 mol/L KCl, the linear range of Cu (II) detection could be extended to 100 mmol/L (6.4 g/L). The reason for the Cu (II) detection limit enhancement was that the reduction of Cu2+ to metallic copper proceeded as two successive single-electron reactions were separated by an intermediate chemical step where the cuprous ion (Cu+) was converted to the dichlorocuprous anion 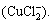 Fe3+ ion interference could be neglected for the detection of Cu (II) using ASDPV once it was lower than 100 mmol/L (5.6 g/L).
Fe3+ ion interference could be neglected for the detection of Cu (II) using ASDPV once it was lower than 100 mmol/L (5.6 g/L).
References
[1] PARDHAN N, NATHSARMA K C, SRINIVASA R K, SUKLA L B, MISHRA B K. Heap bioleaching of chalcopyrite: A review [J]. Minerals Engineering, 2008, 21(5): 355-365.
[2] BAKHTIATI F, ATASHI H, ZIVDAR M, SEYEDBAGHERI S, FAZAELIPOOR M H. Bioleaching kinetics of copper from copper smelters dust [J]. Journal of Industrial and Engineering Chemistry, 2011, 17(1): 29-35.
[3] YIN S H, WU A X, QIU G Z. Bioleaching of low-grade copper sulphides [J]. Transactions of Nonferrous Metals Society of China, 2008, 18(3): 707-713.
[4] KAKSONEN A H, LAVONEN L, KUUSENAHO M, KOLLI A N H, VESTOLA E, PUHAKKA J A, TUOVINEN O H. Bioleaching and recovery of metals from final slag waste of the copper smelting industry [J]. Minerals Engineering, 2011, 24(11): 1113-1121.
[5] NARIN I, SOYLAK M, ELCI L, DOGAN M. Determination of trace metal ions by AAS in natural water samples after preconcentration of pyrocatechol violet complexes on an activated carbon column [J]. Talanta, 2000, 52(6): 1041-1046.
[6] HORVATH Z, LASCHI A, VARGA I, MESZAROS E, MOLNAR A. Determination of trace metals and speciation of chromium ions in atmospheric precipitation by ICP-AES and GFAAS [J]. Talanta, 1994, 41(7): 1165-1168.
[7] GRIEKEN R V. Preconcentration methods for the analysis of water by X-ray spectrometric techniques [J]. Analytica Chimica Acta, 1982, 143: 3-34.
[8] HETTIPATHIRANA T D, SMITH L H, NORRISH K. Simultaneous determination of low parts-per-billion level pb and as in waters using energy-dispersive X-ray fluorescence spectrometry [J]. Appl Spectrosc, 2001, 55(3): 298-306.
[9] MONTERO A A, ESTEVEZ A J R, PADILLA A R. Heavy metal analysis of rainwaters: A comparison of TXRF and ASV analytical capabilities [J]. Journal of Radioanalytical and Nuclear Chemistry, 2007, 273(2): 427-433.
[10] SAUVE S, MCBRIDE M B, HENDERSHOT W H. Ion-selective electrode measurements of copper(II) activity in contaminated soils [J]. Archives of Environmental Contamination and Toxicology, 1995, 29(3): 373-379.
[11] AMACHER M C. Determination of ionic activities in soil solutions and suspensions: Principal limitations1 [J]. Soil Science Society of America Journal, 1984, 48(3): 519-524.
[12] SHAMS E, TORABI R. Determination of nanomolar concentrations of cadmium by anodic-stripping voltammetry at a carbon paste electrode modified with zirconium phosphated amorphous silica [J]. Sensors and Actuators B: Chemical, 2006, 117(1): 86-92.
[13] DESMOND D, LANE B, ALDERMAN J, HILL M, ARRIGAN D W M, FGLENNON J D. An environmental monitoring system for trace metals using stripping voltammetry [J]. Sensors and Actuators B: Chemical, 1998, 48(1-3): 409-414.
[14] HONEYCHURCH K C, HART J P, COWELL D C, ARRIGAN D W M. Voltammetric studies of lead at calixarene modified screen-printed carbon electrodes and its trace determination in water by stripping voltammetry [J]. Sensors and Actuators B: Chemical, 2001, 77(3): 642-652.
[15] ACHTERBERG E P, BRAUNGARDT C. Stripping voltammetry for the determination of trace metal speciation and in-situ measurements of trace metal distributions in marine waters [J]. Analytica Chimica Acta, 1999, 400(1-3): 381-397.
[16] ZOU Z W, JANG A, MACKNIGHT E T, WU P M, DO J, SHIM J S, BISHOP P L, AHN C H. An on-site heavy metal analyzer with polymer lab-on-a-chips for continuous sampling and monitoring [J]. IEEE Sensors Journal, 2009, 9(5): 586-594.
[17] LASCHI S, PALCHETTI I, MASCINI M. Gold-based screen-printed sensor for detection of trace lead [J]. Sensors and Actuators B: Chemical, 2006, 114(1): 460-465.
[18] YANAGIMACHI I, NASHIDA N, IWASA K, SUZUKI H. Enhancement of the sensitivity of electrochemical stripping analysis by evaporative concentration using a super-hydrophobic surface [J]. Science and Technology of Advanced Materials, 2005, 6(6): 671-677.
[19] CRANNY A, HARRIS N R, NIE M Y, WHARTON J A, WOOD R J K, STOKES K R. Sensors for corrosion detection: measurement of copper ions in 3.5% sodium chloride using screen-printed platinum electrodes [J]. IEEE Sensors Journal, , 2012, 12(6): 2091-2099.
[20] MARKOVIC N M, GASTEIGER H A, ROSS P J. Copper electrodeposition on Pt(111) in the presence of chloride and (Bi) sulfate: Rotating ring-Pt(111) disk electrode studies [J]. Langmuir, 1995, 11(10): 4098-4108.
[21] CADLE S H, BRUCKENSTEIN S. Rotating ring-disk study of the underpotential deposition of copper at platinum in 0.5 M hydrochloric acid [J]. Analytical Chemistry, 1971, 43(7): 932-933.
[22] TRNKOVA L, ZERZANKOVA L, DYCKA F, MIKELOVA R, JELEN F. Study of copper and purine-copper complexes on modified carbon electrodes by cyclic and elimination voltammetry [J]. Sensors, 2008, 8(1): 429-444.
[23] MOLINA A, MORALES I. Comparison between derivative and differential pulse voltammetric curves of EC, CE and catalytic processes at spherical electrodes and microelectrodes [J]. International Journal of Electrochemical Science, 2007, 2(5): 386-405.
[24] TAKENO N. Atlas of Eh-pH diagrams [R]. Geological Survey of Japan Report No. 419, 2005.
[25] WANG M S, ZHANG Y, MUHAMMED M. Critical evaluation of thermodynamics of complex formation of metal ions in aqueous solutions III. The system Cu(I,II)-Cl--e at 298.15 K [J]. Hydrometallurgy, 1997, 45(1-2): 53-72.
[26] KEAR G, BARKER B D, WALSH F C. Electrochemical corrosion of unalloyed copper in chloride media�C�CA critical review [J]. Corrosion Science, 2004, 46(1): 109-135.
������������������ұ����ͭ(II)�������
�� ��1,2,3���� ��2�������1���Խ���1
1. �й���ѧԺ �Ϻ�ϵͳ����Ϣ�����о��� ���м������Ϲ���ʵ���ң��Ϻ� 200050��
2. CSIRO Process Science and Engineering, Box 312, Clayton South, Victoria 3169, Australia;
3. �й���ѧԺ��ѧ������ 100049
ժ Ҫ�����ò����������������ʵ������ұ����ͭ(II)Ũ�ȵ�����⡣�������������ϵ�н���ͭ(II)��������Ũ�ȷ�ΧΪ1 ��mol/L~1 mmol/Lʱ��������������������������������������Ũ�ȼ��кܺõ����Զȡ�������ϵ�к���0.2 mol/L KClʱ��ͭ���Ӽ������Է�Χ��1 mmol/L (64 mg/L)��չ��100 mmol/L (6.4 g/L)���ڴ������£�����ͭ���ӵĻ�ԭ��Ϊ���������ĵ�����ת�ƹ��̣��м�̬ΪCu+ �������CuCl-�����⣬��ͭ������ұ����ϵ�У��������������Ӵ��ڣ���˶�ͭ���Ӽ��������Ӹ���Ҳ�������о��������������������Ũ�ȵ���100 mmol/L (5.6 g/L)ʱ�����ͭ���Ӽ��ĸ��ſɺ��ԡ�
�ؼ��ʣ���������������������������ͭ(II)�������Ӹ���
(Edited by Hua YANG)
Foundation item: Projects (2012CB933303, 2011CB707505) supported by the National Basic Research Program of China; Project (2012BAK08B05) supported by the National Key Technology R&D Program of China; Projects (11391901900, 11530700800, 10391901600, 201101042, 11CH-15) supported by Science and Technology Commission of Shanghai Municipality, China; Project supported by Funding from CSIRO CEO Science Leader Program
Corresponding author: Miao CHEN; Tel: +61-3-95458749; E-mail: Miao.Chen@csiro.au
DOI: 10.1016/S1003-6326(14)63098-3

