���±�ţ�1004-0609(2015)-05-1387-07
���õ绯ѧ�����о�LIX84I��ȡCu2+�Ľ���ת�ƹ���
���Ļԣ����øգ��� �ͣ��� 櫣��������������У�����Ԫ
(���ϴ�ѧ ��ѧ����ѧԺ����ɳ 410083)
ժ Ҫ��Ϊ���о�ͭ����ȡ���̣�ͨ������������������绯ѧ�������о�Cu2+��ˮ/1,2-���������������LIX84I����λת�ƹ��̣����Cu2+��LIX84����ɢ����ѧ�����ͽ���ת�ƻ����������������LIX84����Ũ�ȹ���ʱ����ȡ������Cu2+��ˮ��������ɢ������Ĺ���Ϊ���Ʋ��裬����LIX84I�ڽ��淢����λ��Ӧ��ͭ�������������ɢǨ�����л����У���Cu2+���ӹ���ʱ��LIX84I���л���������ɢ�����������Cu2+��λ��ͭ�����Ҳ��������ɢǨ�����л����С�Cu2+��LIX84I����ȡ���������������У����Ͻ������ת�ƺͽ������ת�Ƶ���ɢ������
�ؼ���: Һ/Һ���棻ͭ��ȡ������ת�ƣ��绯ѧ
��ͼ����ţ�TF 811���� ���ױ�־�룺A
Interfacial transfer process of Cu2+ assisted by LIX84I using electrochemical methods
ZHOU Wen-hui, HU Jiu-gang, TANG Hui, LI Ya, FU Ming-bo, SUN Le-xing, CHEN Qi-yuan
(School of Chemical and Chemistry Engineering, Central South University, Changsha 410083, China)
Abstract: In order to study the copper extraction process, based on the electrochemical methods for ion transfer at micro-oil/water interface, the transfer process of Cu2+ assisted by LIX84I at the water/1, 2-dichloroethane interface was investigated. The dynamics parameters of the extraction process and the transfer mechanism of copper(II) were obtained. The results indicate that, when the LIX84I is excess, Cu2+ transfers from aqueous phase to oil/water interface and complexes with LIX84I controlled by a linear diffusion, whereas, the transfer process of LIX84I from the organic phase to oil/water interface and complex with Cu2+ controlled by spherical diffusion when Cu2+ is greatly excess. In both cases, the Cu2+ complexes transfer to the organic phase with a spherical diffusion. The Cu2+ complexes form at interface with an interface complexation transfer and interface dissociative transfer mechanism.
Key words: liquid/liquid interface ; copper extraction; ion transfer; electrochemistry
Һ/Һ������ȡ���̹㷺���������ﻯѧ��ұ����������[1-2]�����ڽ���������ȡ���̣�ͨ��ֻ��ͨ�����ȡ��������к�۷�����������ʱ������߱仯������ֱ���о���ȡ������Ϊ�����ȡ���̵Ķ�̬��Ϣ�������о��߲��õ绯ѧ�����Խ��������ں��Һ/Һ�������ȡ���̽����о����������л���ĸ��迹��Һ/Һ�������ЧӦ�ĸ��ţ����Ի�ÿɿ�������[3]��
����������չ����Һ/Һ����绯ѧ����(Micro-ITIES)����Ч�����л���ĵ���ЧӦ�͵���ЧӦ[4]��������Ӧ�졢�����ȸߵ��ص㣬���ܼ���ģ�ͣ�����Ч���ƴ�ͳ���������ٵ����ݲ�����ȷ�����⣬��ʵ��������ʱ��⣬�ر���������ȡ�����н�������ת�Ƶ��о���Ŀǰ���������о��߲��ô˷�����һ�����ӵĽ���ת�Ʒ�Ӧ�����˹㷺�о�[3, 5-11]��SHAO��[3]�Լ������ˮ/1,2-��������(DCE)�����ϵ�ת�ƹ��̽������о����������4������ת�ƻ���ģ�͡�BEATTIE��[10]��DB18C6���ټ����ӵĽ���ת�ƶ���ѧչ���о���
���ڶ�۽������ӵ���ȡ���̽���ת�Ʒ�Ӧ���������������϶࣬�벻ͬ��������������в��죬����ת�Ƽ���λ���̽�Ϊ���ӣ��䷴Ӧ���̼������о�һֱ�����������⣬������ۺͷ�������Ŀǰ�Դ��ڷ�չ֮�С���DING��[12-14]���������CMPO��TBP������������ȡUO22+��Sr2+�ȷ��������ӵ�ת�ƹ��̽������о�������ͭ������п����ɫ��������ȡ������ʪ��ұ�������ռ������Ҫ��λ[15-20]����YANG��[16]���ò��������ض�Һ����ȡ��[A336][P204]��ȡCe(��)��F(��)�Ķ���ѧ���̽����о����Բ�����ȡ��������ȡ�����еĴ��ʻ��ơ�HEFNY��[17]����·��˹��CYANEX 302��ͭ���̶���ѧ�����о���UNWIN��[19]������Һ�绯ѧ����(MEMED)�����о���Acorga P50��ȡͭ���ӵĶ���ѧ��ز���������Щ����ֻ��ͨ�����ȡ��������ݷ�������ʵ����ȡ���̵�����⡣MASTOURI��[20]������Һ/Һ����绯ѧ������8-�ǻ����ʵ����Pb2+��Cd2+��Zn2+���ؽ������ӿ��ټ�⣬��ͨ���÷����о���ɫ��������ת�ƶ���ѧ����ȴ�ʼ�������
��������ͨ�������绯ѧ����װ�ã�����K+��ת�ƹ������ݼ���װ�õĿ����ԣ���LIX84I����Cu2+��ˮ/1,2-��������(DCE)�����ϵ�ת�ƹ��̽����о����ֱ�������Cu2+��LIX84I��ȡ���������еĶ���ѧ��ɢ��Ϊ��ת�ƻ���������չ����绯ѧ���Է�������ɫ������ȡ��������о��е�Ӧ�á�
1 ʵ��
1.1 ��Ҫ�Լ�������
CHI660D�绯ѧ����վ(�Ϻ���������)��PC-10������(Narishige���ձ�����)��KH-770������ʽ����(HiROX���ձ�����)�����迹�������ñ�(Agilent 34410A����������)������ˮ��(Synergy UV��Millipore)��
�Ȼ���(KCl�����Լ����Ϻ���ҩ����)����ˮ������ͭ(Cu(NO3)2��3H2O�����������Ϻ���ҩ����)��LIX84I(47%��Cognis��˾����)��������-18-��-6 (DB18C6����99.9%��Aladdin��˾����)���Ȼ��Ķ�����(TBACl����������˾����)���ȴ��ı����(KTPBCl��������)��Ϊ�������Լ����ȴ��ı������Ķ�����(TBATPBCl)��KTPBCl�״�/ˮ��Һ(�����2:1)��TBAClˮ��Һ��ϣ��û���Ӧ���ɰ�ɫ�������������˺���ϴ(ϴ����Һ�м�������AgNO3��δ������)��������ͪ�ؽᾧ���Ƶ�[21]��ˮ����л���ֱ��ó���ˮ(18.2 M����cm)��1,2-��������(DCE)���ơ�
1.2 ʵ�鷽��
1.2.1 �缫���Ʊ������
����������PC-10������貣����(�ͺ�BF120���⾶1.2 mm���ھ�0.69 mm������Sutter��˾����)������ͬʱ�õ�������˿�һ�µ�����⡣�����ʵ������������Ƴ��ʺϱ��о��ļ�˽϶̡��ܿڽ�ƽ���ھ��ڼ�����ʮ�IJ����ܡ����������˿ڼ��ⲿ��ò�ֱ���ɨ��羵������������������ͼ1�ɼ���ëϸ�ܹܿ�ƽ�����ھ�Ϊ20.00 ��m���ܱں�7.23 ��m������ⲿ��ò����ǰ�ɴ�ëϸ�ܺ��ע���ɫҺ�壬�Լ�С�����۹�Կ�������Ӱ�졣����ˮ������ͷ��������ëϸ��(�⾶0.52 mm���ھ�0.23 mm)��1 mLע��������ע�룬����ëϸ������ǿ����ЧӦ������Ч���������������ݡ�
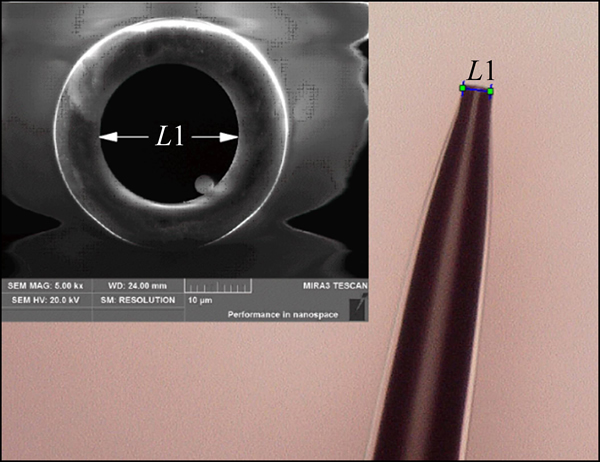
ͼ1 ��ëϸ��������ͼ
Fig. 1 Topography of self-made micropore capillary
1.2.2 �缫���Ʊ�
ˮ��ͨ������Ag/AgCl�缫��Ϊ�����缫������ҵAg/AgCl�缫��ߴ粻�����������о���ϵ�����ƹ����缫���Ʊ��������£����⾶0.2 mm����˿��ĥ����ϴ�ɾ�������0.1 mol/L��KCl��Һ�У���0.3 V�㶨��ѹ����������3 h�������Ƶñ���������ܵ�Ag/AgCl�缫��
���л����У����ڽӴ����ƻ���Һ�л�ȱ仯��Ӱ�죬Ŀǰ����ȷ�ⶨ�л���ĵ�ѹ[22]����ˣ�ʵ����ѡ���ȶ��Ժá����������ظ��ԵIJαȵ缫�Բ����缫���Ʒdz��ؼ����ڷ�ˮ�ܼ���ϵ�ķ������� �У�һ�������˿��˿��Ϊ�αȵ缫����SNOOK��[23]����Ag+/Ag�αȵ缫�ں������������[Pyr4][NTf2]��Һ�вⶨ��ï���ȵ�������ԭ��λ�����������ϵ���нϺ��ȶ��ԡ��ڱ��о��жԱ��������ض��IJαȵ缫����һ���ǽ�Ag/AgCl�缫����10 mmol/L��TBACl��Һ�й����л���TBA+����ѡ���Ե缫���ڶ�����Ag/AgTPBCl������αȵ缫��������Ʊ��������£���ֱ��0.2 mm����˿����ĥ��ϴ����Ϊ���������缫Ϊ��������10 mmol/L��TBATPBCl��Һ�С�1.1 V��ѹֱ����ѹ����������18 h��ʹ�缫������ȸ���һ���ػ�ɫ���塣
1.2.3 �绯ѧ����
���е绯ѧ���Ծ�����CHI660D�绯ѧ����վ������(25��2) ������ɡ�ʵ��������缫��ϵ����װ��ˮ���ëϸ���������л����У��ڲ��������γ�Һ/Һ���档ˮ���в���Ag/AgCl�缫��Ϊ�����缫���л����в���Ag/AgTPBCl�αȵ缫��ʵ�������ϵ�ֱ����£�
���1: Ag/AgCl�缫���ԣ�
Pt | 0.1 mol/L KCl | Ag | AgCl��
���2: TBP+����ѡ���Ե缫���ԣ�
Ag |AgCl | 10 mmol/L TBACl | 1 mmol/L TBATPBCl + 0.6 mmol/L DB18C6 || 100 mmol/L KCl | Ag | AgCl��
���3: Ag/AgTBPCl������缫���ԣ�
Ag |AgTBPCl | 1 mmol/L TBATPBCl + 0.6 mmol/L DB18C6 || 100 mmol/L KCl | Ag | AgCl��
���4: K+ת�Ʒ�Ӧ��
Ag | AgTPBCl | 1 mmol/L TBATPBCl + x mmol/L DB18C6 || y mmol/L KCl | Ag | AgCl��
���5: Cu2+ת�Ʒ�Ӧ��
Ag | AgTPBCl |1 mmol/L TBATPBCl + z mmol/L LIX84I || w mmol/L Cu(NO3)2 | Ag | AgCl��
���6: LIX84Iת�ƿհ�����ԣ�
Ag | AgTPBCl |1 mmol/L TBATPBCl +15 mmol/L LIX84I || 5 mmol/L LiNO3 | Ag | AgCl��
ʵ��װ��ʾ��ͼ��ͼ2��ʾ��
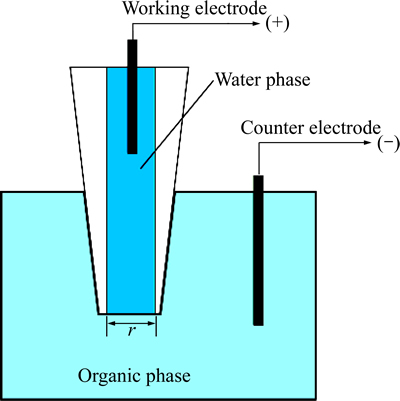
ͼ2 ʵ��װ��ʾ��ͼ
Fig. 2 Scheme of experimental setup
2 ���������
2.1 �������о�
Ϊ��Ӧ��������ת�Ʒ�Ӧ�о��������Ʊ����׳ߴ��Ag/AgCl�缫��Ϊˮ����缫��ͼ3��ʾΪAg/AgCl�缫���л���αȵ缫��ѭ���������ߡ����õ��1��Ӧ��ϵ�����Ƶ�Ag/AgCl�缫����ҵAg/AgCl�缫���жԱȲ��ԣ�ͼ3(a)������������߷���������һ�£�˵�����Ƶ�Ag/AgCl�缫�������ã������ڱ�ʵ�����������ת�Ʋ����о���ͬʱ�����õ��2�͵��3��ϵ�ֱ�����ַ����Ʊ����л���αȵ缫���жԱȲ��ԣ������3(b)��ʾ��ͼ��TPB+����ѡ���Ե缫���ȶ������Ը���Ag/AgTPBCl������缫�����������������Ƚϲ��ˣ�Ag/AgTPBCl������缫���и��õ��ȶ��ԡ�
ͨ���о�DB18C6����K+����ת�ƹ�����֤��ʵ��װ�õĿ����ԣ�������ϵ����4 ��ʾ��ȡ�����Ӵ�ˮ��ת�����л���������Ӵ��л���ת����ˮ��ĵ���Ϊ�������л����������弴0 mmol/Lʱ������ʵ�����0~0.7 V�ĵ��ƴ���Χ�ʺϼ����ӵĽ���ת�ƹ����о�����x=100 mmol/L��y=1 mmol/Lʱ��ͬɨ���ٶ���K+ת�ƹ��̵ķ���������ͼ4��ʾ(xΪDB18C6��Ħ��Ũ�ȣ�yΪKCl��Ħ��Ũ��)���������K+����ת�Ʒ������ɨ�ٵ�ƽ���������ȡ���б�ʼ���K+��ˮ��Һ�е���ɢϵ��Ϊ(2.50��0.01)��10-5 cm2��s-1��������[24]��ֵ�ӽ��������ò���װ�������о����ӽ���ת�ƹ��̾��нϺõĿɿ��ԡ�
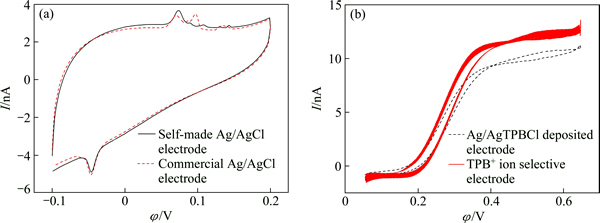
ͼ3 Ag/AgCl�缫���л���αȵ缫��ѭ���������߶Ա�
Fig. 3 Comparison of cyclic voltammogram of Ag/AgCl electrode (a) and reference electrode in organic phase (b)
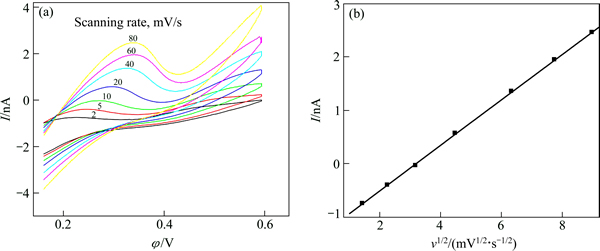
ͼ4 K+��ɢ���ƵĽ�������ת�Ʒ�������
Fig. 4 Voltammogram of K+ transfer at interface (a) and linear relationship between peak current and square root of scanning rate (b)
2.2 Cu2+�Ľ���ת�ƹ���
Ϊȷ����ѡ���ĵ��ƴ���ֻ��Cu2+����ת�ƣ������5�IJ�����ϵ�ֱ����л�������LIX84I(z=0 mmol/L)��ˮ������Cu2+��w=0 mmol/L���������½���ʵ�飬ɨ��Ϊ20 mV/s�������ͼ5��ʾ��
��ͼ5��֪�����л�������LIX84I��������TBATPBCl��Ϊ֧�ֵ����ʱ������ɨ�裬��0.2~0.64 V��Χ��������ת�Ʒ壬˵��������Cu2+����ת�ƣ������Ƴ���0.64 Vʱ�������������ӣ�������ϵ��������������ʱCu2+��ˮ��ת�����л��࣬ͬʱTPBCl-���л���ת����ˮ�ࡣ����ɨ������У������Ƶ���0.2 Vʱ�����������г��ֹյ㣬˵����ϵ���ָ���������ʱNO3-��ˮ��ת�����л��࣬��������TBA+���л���ת����ˮ�ࡣ
��ˮ������Cu2+ʱ����ˮ���м���5 mmol/L LiNO3��Ϊ֧�ֵ�����Ա�֤ˮ��������ǿ�Ƚ��ơ���ͼ5���Կ���������ѹС��0.2 V�ʹ���0.58 Vʱ�����������Ե����ӣ�˵����ϵ�ﵽ����״̬��������������ˮ����Li+���л�����TPBCl-��Ǩ�Ʋ���������������Ϊˮ����NO3-���л�����TBA+��ת������������0.2~0.58 V�ķ�Χ��������ת�Ʒ壬˵���õ��ƴ�����ͭ���Ӻ�����ת�ƣ���LIX84I�Ĵ��ȶԴ�ʵ���о���Ӱ�졣
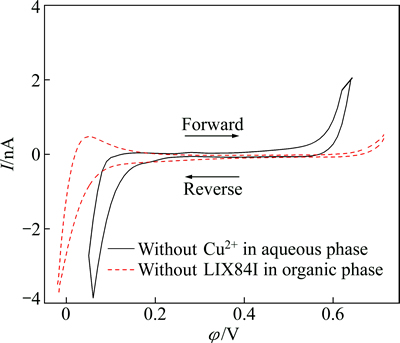
ͼ5 �հ���������ѭ����������
Fig. 5 Cyclic voltammograms of blank experiment sample
���о���������������(z����w)��������ӹ���(w����z)��������Ա��о��÷�Ӧ����ȡ������ʵ������ͼ6��ʾ������ɨ��ʱ������������Ӧ��ȡ��Ӧ����������ȡ����Ϊ��ѧ��Ӧ���ƣ�����������µ�����Ǩ��������ƣ���Ӧ�ķ�����������ҲӦ��ͬ���Ա�ͼ6(a)��(b)��֪����������µķ������������������Ե�����˵���÷�Ӧ���ǻ�ѧ��Ӧ���ơ�
����ëϸ����⼫ϸ��������������Ǩ�Ƴ�������ɢ���������������Ǩ�Ƴ����λ��������ɢ��ëϸ����������Ǩ�ƾ��в��Գ���ɢ��[3]��������ȡ��Ӧ���л������������Ƶ�����ʵ������£�ˮ����Cu2+����Ǩ�����л������������ɢ��������Ӧ���ͷ���������������Ȼͼ6(b)��ʾ����Ϊ��̬�������ߣ��ʸ÷�Ӧ�������л����������ɨ��ʱ�������ͺ�����LIX84I��Cu2+��Ⱦ��и���������ţ�Ǩ������������ͭ�ͺ������ɢ���ơ����������ͭ�ͺ���Ǩ�ƹ��̾�������̬����������������Ӧ������ɢ���ƣ������ù�����ͭ�ͺ��ﲢδ����ˮ�࣬�����ɽ������л���Ǩ�ơ�������ʵ������������Ǩ�ƵIJ��Գ���ɢ����֪���ͺ����ˮ�������Ǩ�ƹ���ӦΪ������ɢ���ɷ�������������
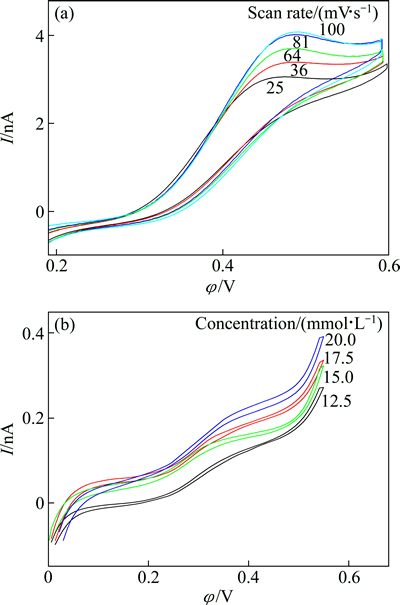
ͼ6 ��ͬɨ����Cu2+��ɢ���ƵĽ�������ת�ƹ���ѭ������ͼ(a)�Ͳ�ͬLIX84Ũ���½�������ת�ƹ��̷���ͼ(b)
Fig. 6 Cyclic voltammograms of Cu2+ transfer across L/L interface in different sweep rate (a) and Cyclic voltammograms of LIX84I transfer across L/L interface in different concentration (b)
���ͼ6��������������֪���������ʱ������ɨ��ʷ��ͷ�����������������ɨ�����̬��������������˵����ȡ������Cu2+��ˮ��������ɢ������Ĺ���Ϊ���Ʋ��裬����LIX84I�ڽ��淢����λ��Ӧ��֮��ͭ�������������ɢ�ӽ���Ǩ�����л��ࡣCu2+����ʱ����ɨ�뷴ɨ��Ϊ��̬�������ߣ�˵��LIX84I���л���������ɢ�����������Cu2+��λ�������Ҳ��������ɢ��ʽ�ɽ���ת�����л��ࡣ�ù��̷���SHAO��[3]����Ľ������ת�ƺͽ������ת�ƻ�����Cu2+��ˮ/1,2-�����������ת�ƹ�����ͼ7��ʾ��
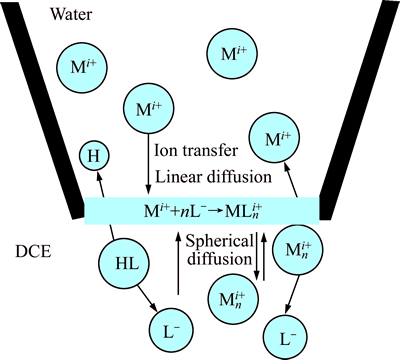
ͼ7 Cu2+��ˮ/1,2-�����������ת�ƹ���ʾ��ͼ
Fig. 7 Scheme of Cu2+ transfer process across water/DCE interface
2.3 ����ת�ƹ��̶���ѧ����
��Cu2+��ɢ��Ϊ���Ʋ���ʱ����ͼ6(a)��ʾ����25~81 mV/s��Χ�ڣ����������ɨ�ٵ����������ɨ������100 mV/sʱ��������������ӣ�����v=81 mV/s��v=100 mV/s�����µķ����������غϣ�����Cu2+��ɢ������ﵽ�ȶ�������ɢ������ͼ8(a)ΪCu2+ɨ��ƽ�����Է����V��ϵͼ����������������ɨ�ٵ�ƽ���������ȡ���ˣ�����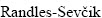 ����(ʽ(1))����ɻ�ý���������ˮ���е���ɢϵ����
����(ʽ(1))����ɻ�ý���������ˮ���е���ɢϵ����
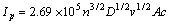 (1)
(1)
ʽ�У�IpΪ�������nΪ���ת������AΪҺ/Һ�������(cm2)��DΪ��ɢϵ��(cm2/s)��vΪɨ���ٶ�(V/s)��cΪŨ��(mol/L)��
�����֪��Cu2+��ˮ��Һ�е���ɢϵ��Ϊ(6.05�� 0.01)��10-6 cm2��s-1��������[25]�н���������K+��ȣ�Cu2+���и�������Ӱ뾶��ˮ���ܡ���ˣ�Cu2+��ˮ���е���ɢϵ������С��K+����ɢϵ����
��LIX84I��ɢ��Ϊ���Ʋ���ʱ����ͼ6(b)��ʾ��Cu2+Ũ��Ϊc=100 mmol/L��ɨ��Ϊ20 mV/s����0.1~0.48 V��Χ�ڣ�ͼ6(b)��4��������ɨ�뷴ɨ���غ��ԽϺã�˵��������ݶ�ϵͳӰ���С��BEATTIE��[10]�������Һ/Һ��������ת�Ʊ��ֵ���ɢ��Ϊ���ڳ������������֮�䣬���ȶ�������ʽ���£�
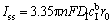 (2)
(2)
ʽ�У�IssΪ��̬������nΪ�������DIΪ������ɢϵ����FΪ�����ڳ�����cIbΪ����ı���Ũ�ȣ�r0Ϊ�뾶��
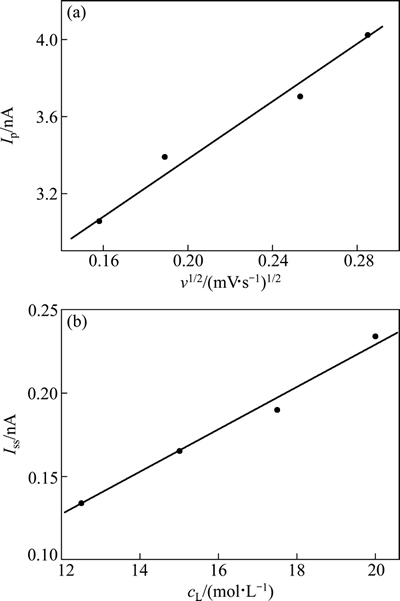
ͼ8 �������ɨ��ƽ������ϵͼ(a)������Ũ�����ȶ�������ϵͼ(b)
Fig. 8 Dependence of peak current on square root of sweep rate (a) and dependence of steady-state current on concentration (b)
LIX84IŨ�����ȶ�������ϵ��ͼ8(b)��ʾ����ʽ(2)�����LIX84��DB18C6��DCE�е���ɢϵ���ֱ�Ϊ(3.19��0.27)��10-10 cm2��s-1��(6.38��0.53)��10-6 cm2��s-1����DCE�ܼ��У�LIX84I����ɢϵ��ԶԶС��DB18C6����ɢϵ�������������LIX84I�Ļ��Žϴ�֧���ϳ����Գ��Խϲ�ռ�λ��ϴ��ԭ����ɵġ�
��Ȼ�й�ͭ������ȡ���̷����ڽ��滹��ˮ����һֱ�����������ۣ�ʵ���ϣ���ȡ����λ������ȡ���������йء�ͨ�������������Եó���Cu2+��LIX84I ��ˮ/1,2-��������(DCE)���������ȡ��Ӧ���˽�����CHAPMAN��[19]�Ķ���ѧ�о����������
3 ����
1) ����Һ/Һ����绯ѧ�����о���Cu2+����ȡ���̡��ڱ�ʵ�������£����������Ӻ������Ǩ�Ƴ��ֲ��Գ���ɢ�����ֱ����ˮ����Cu2+Ϊ���Ʋ�����л�����LIX84IΪ���Ʋ�����������µķ�������������֤ʵ��LIX84I��ȡCu2+�Ĺ��������������У����Ͻ������ת�ƺͽ������ת�ƻ�����������Cu2+��ˮ��Һ�е���ɢϵ��Ϊ(6.05��0.01)��10-6 cm2��s-1��LIX84��DCE�е���ɢϵ��Ϊ(3.19��0.27)��10-10 cm2��s-1��
2) ��Һ/Һ����绯ѧ�о������ܿ�����Ч���о���������Һ/Һ��������ת�ƹ��̵Ķ���ѧ������
REFERENCES
[1] HU J G, CHEN QY, HU HP, YIN ZL. Synergistic extraction of zinc from ammoniacal solutions using ��-diketone mixed with CYANEX923 or LIX84I[J]. Transactions of Nonferrous Metals Society of China, 2012, 22 (5): 1217-1223.
[2] RODRIGUES C E, GON ALVES C B, MARCON E C, BATISTA E A, MEIRELLES A J. Deacidification of rice bran oil by liquid�Cliquid extraction using a renewable solvent[J]. Separation and Purification Technology, 2014, 132: 84-92.
[3] SHAO Y, OSBORNE M, GIRAULT H. Assisted ion transfer at micro-ITIES supported at the tip of micropipettes[J]. Journal of Electroanalytical Chemistry and interfacial Electrochemistry, 1991, 318 (1): 101-109.
[4] �� ��, Ԭ ��. ����/���ܼ����ڵ������ѧ�е�Ӧ��[J]. ������ѧѧ��, 2001, 17(6): 520-525.
SU Bin, YUAN Yi. Application of glass micro and nano meter sized pipettes in electroanalycal chemical[J]. Journal of Analytical Science, 2001, 17(6): 520-525.
[5] �� ݼ, ������, ������, ��Ц��, �� ��, �� ��, ��־ΰ, ��Ԫ��. Һ/Һ����绯ѧ�����չ[J]. �绯ѧ, 2014, 20(3): 234-242.
GU Qin, QIAO Yong-hui, ZHU Xin-yu, YIN X, ZHANG X, CHEN Y, ZHU Z, SHAO Y. Electrochemistry at liquid/liquid interfaces and its recent progress[J]. Jouranl of Electrochemistry, 2014, 20(3): 234-242.
[6] QIAO Y, ZHANG B, ZHU X, JI T, LI B, LI Q, CHEN E, SHAO Y. Facilitated ion transfers at the micro�\water/1, 2�\dichloroethane interface by crown ether derivatives[J]. Electroanalysis, 2013, 25(4): 1080-1084.
[7] STOCKMANN T J, MONTGOMERY A-M, DING Z. Determination of alkali metal ion transfers at liquid|liquid interfaces stabilized by a micropipette[J]. Journal of Electroanalytical Chemistry, 2012, 684: 6-12.
[8] STOCKMANN T J, MONTGOMERY A-M, DING Z. Correlation of stoichiometries for Rb+ extraction determined by mass spectrometry and electrochemistry at liquid|liquid interfaces[J]. Analytical chemistry, 2012, 84(14): 6143-6149.
[9] VEL ZQUEZ-MANZANARES M. Fundamentals and applications in electrochemistry of liquid-liquid interfaces[J]. Procedia Chemistry, 2014, 12: 100-107.
[10] BEATTIE P, DELAY A, GIRAULT H. Investigation of the kinetics of assisted potassium ion transfer by dibenzo-18-crown-6 at the micro-ITIES by means of steady-state voltammetry[J]. Journal of Electroanalytical Chemistry, 1995, 380(1): 167-175.
[11] ղ��ƽ, �����. ������ 18 �� 6 �ٽ���������Һ-Һ�����ϵ�Ǩ�Ʒ�Ӧ[J]. �绯ѧ, 2002, 8(3): 252-256.
ZHAN Dong-ping, WU Bing-liang. Facilitated K+ ion transfer by DB18C6 across the micro liquid/liquid interface[J]. Journal of Electrochemistry, 2002, 8(3): 252-256.
[12] STOCKMANN T J, LU Y, ZHANG J, GIRAULT H H, DING Z. Interfacial complexation reactions of Sr2+ with octyl (phenyl)-N,N-diisobutylcarbamoylmethylphosphine oxide for understanding its extraction in reprocessing spent nuclear fuels[J]. Chemistry, 2011, 17(47): 13206-13216.
[13] STOCKMANN T J, DING Z. Uranyl ion extraction with conventional PUREX/TRUEX ligands assessed by electroanalytical chemistry at micro liquid/liquid interfaces[J]. Analytical Chemistry, 2011, 83(19): 7542-7549.
[14] STOCKMANN T J, ZHANG J, MONTGOMERY A M, DING Z. Electrochemical assessment of water| ionic liquid biphasic systems towards cesium extraction from nuclear waste[J]. Analytica chimica acta, 2014, 821: 41-47.
[15] ���Ӻ�, ������, Ƚ ӯ, ����Ƽ, ����Ԫ. ��λ���-��ͪ��LIX84�����ȡ���Ӱ�����Һ����ȡͭ[J]. �й���ɫ����ѧ��, 2011, 21(5): 1171-1177.
XIANG Yan-hong, YIN Zhou-lan, RAN Ying, HU Hui-ping, CHEN Qi-yuan. Solvent extraction of copper from ammonia solutions by sterically hindered ��-diketone and LIX 84[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(5): 1171-1177.
[16] YANG Hua-ling, CHEN Ji, ZHANG Dong-li, WEI Wei, CUI Hong-min, LIU Yu. Kinetics of cerium (IV) and fluoride extraction from sulfuric solutions using bifunctional ionic liquid extractant (Bif-ILE)[A336][P204][J]. Transactions of Nonferrous Metals Society of China, 2014, 24(6): 1937-1945.
[17] EL HEFNY N. Kinetics and mechanism of extraction of Cu (II) by CYANEX 302 from nitrate medium and oxidative stripping of Cu (I) using Lewis cell technique[J]. Chemical Engineering and Processing: Process Intensification, 2010, 49(1): 84-90.
[18] XING Peng, WANG Cheng-yan, XU Sheng-ming, JU Zhong-jun. Kinetics of cobalt (II) extraction from sulfate aqueous solution by sodium salt of di-decylphosphinic acid (DDPA)[J]. Transactions of Nonferrous Metals Society of China, 2013, 23(2): 517-523.
[19] ZHANG J, CHAPMAN D, SLEVIN C J, UNWIN P R. Investigation of cupric ion extraction kinetics in a two-phase organic/water system using microelectrochemical measurements at expanding droplets (MEMED)[J]. Journal of Electroanalytical Chemistry, 2002, 538: 277-283.
[20] MASTOURI A, PEULON S, BELLAKHAL N, CHAUSS A. M (II) transfer across a liquid-liquid microinterface facilitated by a complex formation with 8-Hydroxyquinoline: Application to quantification of Pb (II), Cd (II) and Zn (II) alone or in mixture in effluents[J]. Electrochimica Acta, 2014, 130: 818-825.
[21] Ԭ ��, �� ��, ��Ԫ��. Ӧ�ò�����/���ܺ�ѭ���������о��ڷƿ�������������ˮ/1, 2��������������ת�ƹ���[J]. �ߵ�ѧУ��ѧѧ��, 2001, 22(11): 1819-1823.
YUAN Yi, SU Bin, SHAO Yuan-hua. Investigation of proton transfer across the water/1,2-dichloroethane interface by o-Phenant hroline using glass micro/nano-pipets and cyclic voltammetry[J]. Chemical Journal of Chinese Universities, 2001, 22 (11): 1819-1823.
[22] HAPIOT P, LAGROSTC. Electrochemical reactivity inroom-temperature ionic liquids[J]. Chemical Reviews, 2008, 108(7): 2238-2264.
[23] SNOOK G, BEST A, PANDOLFO A, HOLLENKAMP A. Evaluation of a Ag�OAg+ reference electrode for use in room temperature ionic liquids[J]. Electrochemistry Communications, 2006, 8(9): 1405-1411.
[24] ��ȫ��, �缫���̶���ѧ����[M]. ����: ��ѧ������, 2002.
ZHA Quan-xing. The Introduction of ectrode ocesses knetics[M]. Beijing: Science Press, 2002.
[25] ZHANG J, UNWIN P R. Microelectro-chemical measurements at expanding droplets (MEMED): Investigation of cupric ion stripping kinetics in a two-phase oil/water system[J]. Physical Chemistry Chemical Physics, 2000, 2 (6): 1267-1271.
(�༭ ������)
������Ŀ�������ص�����о���չ�ƻ�������Ŀ(2014CB643401)��������Ȼ��ѧ����������Ŀ(51134007��51304244)���й���ʿ���ѧ����������Ŀ(2014M552152)�����ϴ�ѧ��ʿ�����������Ŀ
�ո����ڣ�2014-09-22�������ڣ�2015-01-22
ͨ�����ߣ�����Ԫ�����ڣ���ʿ���绰��0731-88877478��E-mail��cqy@csu.edu.cn

