文章编号:1004-0609(2008)06-1164-07
氟铝酸盐离子团簇微环境与拉曼光谱表征
肖 丽,尤静林,蒋国昌
(上海大学 材料科学与工程学院,上海 200072) Z
摘 要:对碱金属氟铝酸盐系几种典型晶体的拉曼光谱进行研究,并用量子化学从头计算方法以氟铝酸盐为对象构建一些超分子形式的离子团簇,采用Restricted Hartree-Fock (RHF)自洽场方法和基组6-31G(d)对其进行优化,在相同基组和方法条件下,计算其拉曼振动频率及其拉曼活性,将实验结果与计算值进行对比分析。结果表明,拉曼位移能够准确反映微观环境的差异或超分子聚集数,即随超分子聚集数的增加,Al―F对称伸缩振动频率的计算值越接近实验值。
关键词:氟铝酸盐;离子团簇;微环境;拉曼光谱
中图分类号:O 657.37
Characteristic Raman spectra of micro environment of fluoroaluminate structural units
XIAO Li, YOU Jing-lin, JIANG Guo-chang
(School of Materials Science and Engineering, Shanghai University, Shanghai 200072, China)
Abstract: The Raman spectra of several typical crystals of alkali fluoroaluminate system were studied. Ab initio calculation of quantum chemistry was used to construct several super molecule model clusters. Restricted Hatree-Fock method (RHF) with the basis set of 6-31G(d) was applied to optimize the geometry of fluoroaluminate model clusters. The wavenumbers of Raman-active modes and Raman optical activity (ROA) were calculated under the same method with the same base set after geometric optimization. The calculation results were compared with the experimental data. The results show that the delicate variation of micro-environment or super molecular number can be correctly resolved by Raman shift. That is, the calculated Raman wavenumber of Al―F symmetric stretching vibration is much closer to experimental data when super molecular number gradually increases.
Key words: fluoroaluminate; model clusters; micro environment; Raman spectra
碱金属氟铝酸盐在铝工业生产中的应用引起人们广泛的兴趣,如冰晶石(Na3AlF6),其熔体为离子化合物,具有很强的溶解Al2O3的能力[1],是霍尔-埃鲁法,即冰晶石-氧化铝熔盐电解法生产铝的熔剂[2]。
物质的微观结构决定其宏观物理和化学性能,氟铝酸盐内部微结构决定着其宏观性能。因此,了解氟铝酸盐的微观结构,可以深入了解铝电解体系熔盐物理化学性质的变化规律,正确认识熔盐电解的电极过程,了解铝电解机制,以便优化和改进生产过程,为提升和改造传统产业提供理论基础。目前,存在几种从体系的微观结构单元出发研究其宏观性质的实验手段,而近几年来发展起来的拉曼光谱技术,已成为除X射线衍射和核磁共振等方法以外的研究物质结构的主要手段[3-4]。SOLOMONS等[5]对冰晶石熔体和晶体的拉曼光谱进行了研究,并对谱图的各个谱峰进行指认,指出固相状态的冰晶石配位数为6,即 ,其峰值为554 cm-1。BROOKER等[6]研究了固体和熔融状态的LiF/NaF/KF以及固体Na3AlF6,K3AlF6和K2NaAlF6的六氟铝酸盐离子的拉曼光谱,指出孤立的ν1振动模式峰的最大值随晶体堆积的不同有很大的变化。随着计算机技术的迅速发展,量子化学计算作为一种重要的研究手段在氟铝酸盐的结构研究中也取得了很大进展[7-9]。侯怀宇等[10]用分子动力学的方法模拟计算(NaF)x(AlF3)1-x熔盐体系在1 323K时的结构。SPOLITI等[11]对具有Td对称性的
,其峰值为554 cm-1。BROOKER等[6]研究了固体和熔融状态的LiF/NaF/KF以及固体Na3AlF6,K3AlF6和K2NaAlF6的六氟铝酸盐离子的拉曼光谱,指出孤立的ν1振动模式峰的最大值随晶体堆积的不同有很大的变化。随着计算机技术的迅速发展,量子化学计算作为一种重要的研究手段在氟铝酸盐的结构研究中也取得了很大进展[7-9]。侯怀宇等[10]用分子动力学的方法模拟计算(NaF)x(AlF3)1-x熔盐体系在1 323K时的结构。SPOLITI等[11]对具有Td对称性的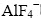 及等的优化构型和振动频率进行计算,用HF-SCF和MP2计算方法对Li+和Na+与这些阴离子的相互作用进行研究。AKDENIZ等[12]研究用碱金属离子来稳定复杂阴离子(AlF5)2-的理论,计算了MnAlFn+3(M=Li, Na或K和n=2或3)团簇的内聚能,结构和键长以及在不同MnAlFn+3(M=Li, Na或K)团簇中的、AlF2-5和AlF4-呼吸振动的频率。这些研究对熔体和玻璃体系有重要的意义,但搭建的团簇体系比较小,难以模拟真实的微环境。本文作者对碱金属氟铝酸盐系几种典型晶体的拉曼光谱进行了研究。针对有限尺度晶体的有限晶体结构来模拟其真实的微观环境,采用Gassian03W量子化学计算软件对氟铝酸盐离子团簇的构型进行空间结构优化,计算拉曼振动频率及其拉曼活性,为高温熔体解谱的定量分析奠定基础。
及等的优化构型和振动频率进行计算,用HF-SCF和MP2计算方法对Li+和Na+与这些阴离子的相互作用进行研究。AKDENIZ等[12]研究用碱金属离子来稳定复杂阴离子(AlF5)2-的理论,计算了MnAlFn+3(M=Li, Na或K和n=2或3)团簇的内聚能,结构和键长以及在不同MnAlFn+3(M=Li, Na或K)团簇中的、AlF2-5和AlF4-呼吸振动的频率。这些研究对熔体和玻璃体系有重要的意义,但搭建的团簇体系比较小,难以模拟真实的微环境。本文作者对碱金属氟铝酸盐系几种典型晶体的拉曼光谱进行了研究。针对有限尺度晶体的有限晶体结构来模拟其真实的微观环境,采用Gassian03W量子化学计算软件对氟铝酸盐离子团簇的构型进行空间结构优化,计算拉曼振动频率及其拉曼活性,为高温熔体解谱的定量分析奠定基础。
1 实验
1.1 实验装置
实验所用拉曼光谱仪为法国 Jobin-Yvon公司的LabRAM HR-800型激光共焦Raman光谱仪,并配置了增强型电荷耦合探测器(ICCD, Intensified Charge Coupled Device),集成了空间分辨和累积时间分辨耦合测量机制。激光光源采用Coherent Compass 501QM-VD型半导体固体脉冲激光器,并配置了时间分辨同步探测系统,可与脉冲激光同步工作,在高温测量中提高信噪比[13]。本实验所用激光波长为532 nm,激光平均功率约0.22 W,脉冲频率为10 kHz,狭缝宽度300 μm,脉冲时间10 ns,扫描次数200 次,扫描波数范围为100~800 cm-1。
1.2 样品制备
K2NaAlF6晶体是由无水NaF,KF和AlF3制得。将化学纯无结晶水试剂NaF在423 K温度下烘干,AlF3在573 K退火12 h去除吸收的水分,KF×2H2O在773 K脱水得到无水KF [14],然后按照一定的摩尔比例混合,置于玛瑙研钵中研磨约30 min,以确保粉末充分混合均匀。把混匀的粉末装入铂金坩埚中,置于高温电炉内,逐渐升温至熔点以上,恒温1 h待试样完全熔融后,以一定的速率缓慢降到室温,便得到K2NaAlF6晶体。Na3AlF6和K3AlF6是化学纯无结晶水试剂,在423 K温度下烘干即可。
2 结果与讨论
2.1 几种氟铝酸盐晶体的拉曼光谱
图1所示为几种氟铝酸盐晶体的拉曼光谱。由图可知,各拉曼峰的频率与BROOKER等[6]的测量结果基本吻合。各谱中的最强峰都是由于结构单元中ALF3-6铝氟键的对称伸缩振动产生的拉曼频移,Na3AlF6、K3AlF6和K2NaAlF6这3种晶体的对称伸缩振动峰分别在552、545和561 cm-1处。由此可见,由于阳离子的不同,中铝氟键的对称伸缩振动产生的拉曼频移略有不同。图1所示的Na3AlF6谱中342和394 cm-1属于铝氟键的反对称弯曲振动和反对称伸缩振动,K3AlF6谱中324 cm-1属于铝氟键的反对称伸缩振动,K2NaAlF6谱中327 cm-1属于铝氟键的反对称伸缩振动。
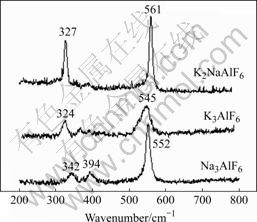
图1 室温下Na3AlF6, K2NaAlF6 和 K3AlF6晶体的实验Raman光谱
Fig.1 Experimental Raman patters of Na3AlF6, K2NaAlF6 and K3AlF6 crystals at room temperature
2.2 量子化学从头计算
在氟铝酸盐系中,八面体除了以长链和环的方式聚集外,在六氟铝酸盐体系里是以孤立的八面体聚集。参考Na3AlF6(a)、K3AlF6(b)和K2NaAlF6(c)这3种晶体的结构,图2所示为计算所搭建的团簇模型的构型图。本工作中使用Gassian03W量子化学软件,采用Restricted Hartree-Fock自洽场方法,在6-31G(d)基组水平上对图2所示团簇结构单元进行了空间结构优化,并使用相同基组和方法计算其拉曼振动频率及拉曼活性。

图2 从头计算中采用的团簇模型的构型图
Fig.2 Diagrams of model clusters used for calculations: element labels of (a-2) to (a-4) are same as in diagram of (a-1); element labels of (b-2) to (b-4) are same as in diagram of (b-1); Element labels of (c-2) to (c-4) are same as in diagram of (c-1); 1, 2, 3 and 4 respectively represent different numbers of super molecule
a-1、b-1和c-1这3种离子团簇结构体系小,离子团簇中包含的结构单元数目少,只考虑结构单元本身的变化,并且仅凭自身平衡的几个阳离子围绕在阴离子团簇周围,离子团簇结构参数(键长和键角等)发生改变的自由程度较大,与实际晶体的微观环境存在较大差异。
针对a-1、b-1和c-1这3种离子团簇结构较小,阴离子基团周围配位的阳离子数不足以及与实际晶体的微观环境存在较大差异的问题,采取向原构型a-1、b-1和c-1中分别加入处于近邻位置的a-1、b-1和c-1结构单元的方法来增大离子团簇的体系规模,以考察离子团簇的体系大小对拉曼特征峰的影响。表1所列为Na3AlF6、K3AlF6 和K2NaAlF6这3种晶体实验测得的拉曼频率与图2所列的团簇结构单元的计算频率。
首先,以Na3AlF6晶体为对象,构建了几种超分子形式的团簇进行计算,计算得到的拉曼光谱与实验结果相比较时,需要对温度与频率进行校正[15],校正后的结果如图3所示。由图3可知,超分子团簇a-1的计算值与实验值相差较大,且其计算拉曼光谱在低频区出现严重的谱峰分裂现象,这是由于低聚体时,分子的对称性较差,以及拉曼谱峰的简并度较低。二聚体时,聚集体数目增加,分子的对称性增强,谱峰的简并度提高,谱峰分裂现象消失,且计算得到的谱图与实验测得的谱图峰形较吻合;四聚体时,计算谱图与实验谱图接近。

图3 Na3AlF6晶体的实验拉曼光谱和相应团簇经校正的计算拉曼光谱
Fig.3 Experimental Raman spectras of Na3AlF6 crystal and reduced calculated Raman spectra of corresponding model clusters
表1中,二聚体时,铝氟键的对称伸缩振动频率由474 cm-1增加到504 cm-1;三聚体时,频率增加到519 cm-1;四聚体时,频率继续增加到529 cm-1。以上结果表明,随超分子数的增加铝氟键的对称伸缩振动频率的计算值与实验值接近。这是因为从单聚体到四聚体,单个铝氟八面体受周围微观环境的牵制,应力或束缚愈大而导致Al―F键的对称伸缩振动频率发生蓝移。ZHU等[16]报道的随非晶金刚石薄膜厚度的增加,即残余压应力的降低,则其对应的Raman 峰位向低频移动。YOU等[17]为阐明拉曼振动的微观应力作用机制定义的硅氧四面体应力指数SIT亦有类似的变化。以上分析说明本模拟对Al―F键的对称伸缩振动频率随超分子数目增加发生蓝移现象的解释是准确可靠的。
参照冰晶石的晶体结构,1个孤立的ALF3-6八面体周围微环境中有8个孤立的八面体。根据上述分析,将铝氟键的对称伸缩振动频率的实验值(a)与不同超分子数的铝氟键的对称伸缩振动频率的计算值(a-1~a-4)进行了拟合,拟合的表达式为lg(554.45-y)= lg119.13+xlg 0.67,结果如图4所示。由图可知,随超分子数的增加,铝氟键对称伸缩振动频率的计算值与实验值接近,当有9个超分子聚集时,与晶体结构的实际微环境一致时,计算值将会与实验值吻合。
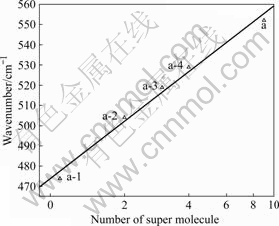
图4 Na3AlF6晶体Al―F的对称伸缩振动实验和计算频率与超分子数的相关关系
Fig.4 Number of super molecule dependent experimental and calculated wavenumbers of Al―F symmetrical stretching vibration with Na3AlF6 considered
K3AlF6晶体的实验拉曼光谱和相应团簇经校正的计算拉曼光谱如图5所示。由图可知:超分子团簇b-1的计算拉曼光谱在低频区同样出现了谱峰分裂的现象。超分子聚集数为2时,谱峰分裂现象消失;增加到3时,团簇b-2低频区的两个峰简并,且计算得到的谱图与实验测得的谱图峰形较吻合; 当达到4时,其拉曼光谱却变化较小,保持相对稳定的峰形。

图5 K3AlF6晶体的实验拉曼光谱和相应团簇经校正的计算拉曼光谱
Fig.5 Experimental Raman spectras of K3AlF6 crystal and reduced calculated Raman spectra of corresponding model clusters
表1 几种晶体拉曼振动模式的实验频率和计算频率
Table 1 Experimental and calculated wavenumbers of Raman active modes of several fluoroaluminate crystals
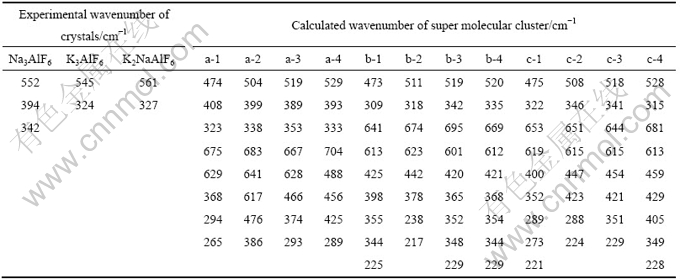
由表1计算得到的拉曼频率可以发现,K3AlF6晶体与Na3AlF6晶体有相同的变化规律。当超分子聚集数由1变到2,3和4时,铝氟键的对称伸缩振动频率由473 cm-1增大到511,519和520 cm-1。ABAKUMOV等[14]曾提到,对于K3AlF6晶体的结构信息是非常有限的,目前只是用与其结构相似的K2NaAlF6的晶体结构来描述。参照K2NaAlF6的晶体结构,发现1个孤立的八面体周围微环境中有12个孤立的八面体。根据上述分析,将铝氟键的对称伸缩振动频率的实验值(b)与不同超分子数的铝氟键的对称伸缩振动频率的计算值(b-1~b-4)进行拟合,拟合的表达式为lg(542.25-y) = lg 110.28+xlg 0.60,结果如图6所示。由图可知,随超分子数的增加,铝氟键对称伸缩振动频率的计算值与实验值接近,当13个超分子聚集,与晶体结构的实际微环境一致时,计算值将会与实验值比较吻合。

图6 K3AlF6晶体Al―F的对称伸缩振动实验和计算频率与超分子数的相关关系
Fig.6 Number of super molecule dependent experimental and calculated wavenumbersof Al―F symmetrical stretching vibration with K3AlF6 considered
K2NaAlF6晶体的实验拉曼光谱和相应团簇经校正的计算拉曼光谱如图7所示。由图可知,其计算拉曼光谱随超分子数增加的变化规律与Na3AlF6晶体的非常相似,在此不再详细分析。
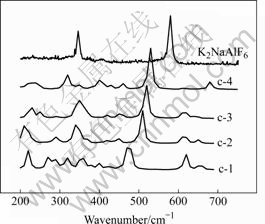
图7 K2NaAlF6晶体的实验拉曼光谱和相应团簇经校正的计算拉曼光谱
Fig.7 Experimental Raman spectras of K2NaAlF6 crystal and reduced calculated Raman spectra of corresponding model clusters
由表1计算得到的拉曼频率可以发现,K2NaAlF6晶体与Na3AlF6和K2NaAlF6晶体的变化规律相同。当超分子聚集数由1变化到2,3和4时,铝氟键的对称伸缩振动频率由475 cm-1增大到508,518和528 cm-1。
参照K2NaAlF6的晶体结构,1个孤立的ALF3-6八面体周围微环境中有12个孤立的ALF3-6八面体。将铝氟键的对称伸缩振动频率的实验值(c)与不同超分子数的铝氟键的对称伸缩振动频率的计算值(c-1~c-4)进行拟合,拟合的表达式为lg (561.62-y) = lg 117.74+ xlg 0.71,结果如图8所示。由图变化趋势与K3AlF6晶体类似,但要优于K3AlF6晶体的拟合结果。
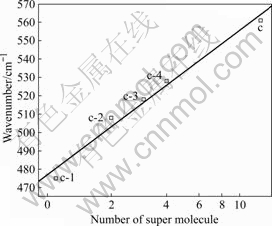
图8 K2NaAlF6晶体Al―F的对称伸缩振动实验和计算频率与超分子数的相关关系
Fig.8 Number of super molecule dependent experimental and calculated wavenumbers of Al―F symmetrical stretching vibration with K2NaAlF6 considered
Na3AlF6,K3AlF6和K2NaAlF6 这3种晶体的铝氟键的对称伸缩振动频率的计算值随超分子数的增加而增大,这表明超分子结构单元间的相互作用非常明显。YOU等[18]针对二元钠硅酸盐的计算结果亦有类似的变化。以上分析说明本文对氟铝酸盐晶体的模拟计算数据是可靠的。
由以上分析可知,拉曼位移能够准确反映微观环境的不同或超分子聚集数。但是,无论对Na3AlF6晶体,K3AlF6晶体还是K2NaAlF6晶体,由于超分子结构的设计还未尽合理,其影响机制还有待进一步的研究。此外本计算模拟是针对有限尺度晶体的有限晶体结构,特别是当体系处于高温熔融状态时,它有非常重要的意义,有待以后进行更加深入的研究。
3 结论
1) 实测了碱金属氟铝酸盐系几种典型晶体的拉曼光谱,解释了各谱峰的振动模式。使用量子化学从头计算方法模拟计算了氟铝酸盐超分子离子团簇。
2) 拉曼位移能够准确反映微观环境的差异或超分子聚集数,即随超分子聚集数的增加,铝氟键对称伸缩振动频率的计算值越接近实验值。
REFERENCES
[1] 冯乃祥. 铝电解[M]. 北京: 化学工业出版社, 2006: 1-2.
FENG Nai-xiang. Aluminum elecetrolysis[M]. Beijing: Chemical Industry Press, 2006: 1-2.
[2] AUGUSTE F, TKATCHEVA O, MEDIAAS H, ?STVOLD T, GILBERT B. The dissociation of fluoroaluminates in FLiNaK and CsF-KF molten mixtures: A Raman spectroscopic and solubility study[J]. Inorganic Chemistry, 2003, 42(20): 6338-6344.
[3] 蒋国昌, 尤静林, 余丙鲲, 黄世萍. 高温Raman光谱测试技术进展[J]. 光谱学与光谱分析. 2000, 20(2): 206-209.
JIANG Guo-chang, YOU Jing-lin, YU Bing-kun, HUANG Shi-ping. Developments of high temperature Raman spectroscopic techniques[J]. Spectroscopy and Spectral Analysis, 2000, 20(2): 206-209.
[4] YOU Jing-lin, JIANG Guo-chang, XU Kuang-di. High temperature Raman spectra of sodium disilicate crystal, glass and its liquid[J]. J Non-Cryst Solids, 2001, 282(1): 125-131.
[5] SOLOMONS C, CLARKE J H R, BOCKRIS J O’ M. Identification of the complex ions in liquid cryolite[J]. J Chem Phys, 1968, 49(1): 445-449.
[6] BROOKER M H, BERG R W, von BARNER J H, BJERRUM N J. Raman study of the hexafluoroaluminate ion in the solid and molten FLiNaK[J]. Inorg Chem, 2000, 39(16): 3682-3689.
[7] AKDENIZ Z, ?I?EK Z, TOSI M P. Theoretical evidence for the stability of the (AlF2-5)complex anion[J]. Chemical Physics Letters, 1999, 308: 479-485.
[8] SCHOLZ G, CURTISS L A. Ab initio MO calculations of high temperature gaseous fluorine complexes MAlF4(M= H, Li or Na): a comparative study using different basis sets[J]. Journal of Molecular Structure(Theohem), 1992, 258: 251-260.
[9] GUTSEV G L, JENA P, BARTLETT R J. Structure and stability of BF3*F and AlF3*F superhalogens[J]. J Chemical Physics Letters, 1998, 292: 289-294.
[10] 侯怀宇, 谢 刚, 陈书荣, 张雄飞. NaF2-AlF3系熔盐结构的分子动力学计算[J]. 中国有色金属学报, 2000, 10(6): 914-918.
HOU Huai-yu, XIE Gang, CHEN Shu-rong, ZHANG Xiong-fei. Structure of molecular dynamics simulated NaF-AlF3 melt[J]. The Chinese Journal of Nonferrous Metals, 2000, 10(6): 914-918.
[11] SPOLITI M, SANNA N, MARTINO V D. Ab initio study on the MBF4 and MAlF4 molecules[J]. Journal of Molecular Structure (Theochem), 1992, 258: 83-107.
[12] AKDENIZ Z, ?I?EK Z, KARAMAN A, PASTORE G, TOSI M P. A theoretical study of the stabilization of the (AlF2-5) complex anion by alkali counterions[J]. Z Naturforsch, 1999, 54A: 575-578.
[13] 尤静林. 高温拉曼光谱创新技术、光谱计算和在无机化合物微结构研究中的应用[D]. 上海: 上海大学, 2006: 56-63.
YOU Jing-lin. Novel high temperature Raman spectroscopic techniques, spectral calculation and their application in micro-structure study of inorganic materials[D]. Shanghai: Shanghai University, 2006: 56-63.
[14] ABAKUMOV A M, ROSSEL M D, ALEKSEEVA A M, VASSILIEV S Y, MUDREZOVA S N, TENDELOO G V, ANTIPOV E V. Phase transitions in K3AlF6[J]. Journal of Solid State Chemistry, 2006, 179: 421-428.
[15] YOU Jing-lin, JIANG Guo-chang, HOU Huai-yu, WU Yong-quan, XU Kuang-di. Qutantum chemistry study on superstructure and Raman spectra of binary sodium silicates[J]. Journal of Raman Spectroscopy, 2005, 36: 237-249.
[16] ZHU Jia-qi, HAN JIE-cai, LIU Ai-ping, MENG Song-he, JIANG Chun-zhu. Mechanical properties and Raman characterization of amorphous diamond films as a function of film thickness[J]. Surface & Coatings Technology, 2007, 201: 6667-6669.
[17] YOU Jing-lin, JIANG Guo-chang, Hou Huai-yu, CHEN Hui, WU Yong-quan, XU Kuang-di. An ab-initio calculation of Raman spectra of binary sodium silicates[J]. Chin Phys Lett, 2004, 21(4): 640-643.
[18] YOU Jing-lin, JIANG Guo-chang, CHEN Hui, XU Kuang-di. Raman spectra and structure study of silicate glasses and their liquids[J]. Rare Metals, 2006, 25(5): 431-436.
基金项目:国家自然科学基金资助项目(50334040,50334050;40203001,50472104);上海市教育委员会创新资助项目(08YZ11)
收稿日期:2007-09-20;修订日期:2008-01-09
通迅作者:尤静林,教授,博士;电话:021-56331482;E-nail:jlyou@staff.shu.edu.cu
(编辑 龙怀中)

