


Trans. Nonferrous Met. Soc. China 22(2012) 1197-1202
First principles study of electronic structure, chemical bonding and elastic properties of BiOCuS
PAN Liu-xian1, XIA Qing-lin2, YE Shao-long3, DING Ning1, LIU Zi-ran1
1. School of Electrical and Information Engineering, Hunan International Economics University,Changsha 410205, China;
2. School of Physics and Electronics, Central South University, Changsha 410083, China;
3. School of Metallurgical Science and Engineering, Central South University, Changsha 410083, China
Received 17 January 2012; accepted 19 April 2012
Abstract: The electronic structures, chemical bonding and elastic properties of the tetragonal phase BiOCuS were investigated by using density-functional theory (DFT) within generalized gradient approximation (GGA). The calculated energy band structures show that the tetragonal phase BiOCuS is an indirect semiconductor with the calculated band gap of about 0.503 eV. The density of states (DOS) and the partial density of states (PDOS) calculations show that the DOS near the Fermi level is mainly from the Cu-3d state. Population analysis suggests that the chemical bonding in BiOCuS has predominantly ionic character with mixed covalent-ionic character. Basic physical properties, such as lattice constant, bulk modulus, shear modulus, elastic constants, were calculated. The elastic modulus and Poisson ratio were also predicted. The results show that tetragonal phase BiOCuS is mechanically stable and behaves in a ductile manner.
Key words: BiOCuS; first principles; electronic structures; chemical bonding; elastic properties
1 Introduction
Since the discovery of superconductivity in LaOFeP, LaNiOP and LaFeAsO(F) [1-3], the quaternary oxypnictides and oxysulfides layered compounds with the ZrCuSiAs-type structure (space group P4/nmm, Z=2) have received considerable attention because of their interesting electrical, optical and magnetic properties [4-8].
BiCuOS, which is isostructural to the layered rare-earth oxysulfides LnCuOS (Ln=La-Eu), is an indirect gap semiconductor with band gap of about 1.1 eV in experimental investigation [7] or about 0.48 eV in theoretical calculation [9]. KUSAINOVA et al [10] synthesized BiOCuS by a high-temperature reaction in evacuated sealed quartz ampoules. SHEETS et al [8] synthesized BiCuOS by a single-step hydrothermal reaction at low temperature (250 °C) and pressure (<2?106 Pa). They studied the optical properties of BiCuOS with diffuse reflectance. The absorption spectrum of BiCuOS is high across the entire visible region (2-3 eV), indicating that the optical band gap of BiCuOS is much smaller (<1.5 eV). HIRAMATSU et al [7] prepared BiOCuS samples by solid-state reaction method, and estimated the band gap of BiCuOS (about 1.1 eV) by diffuse reflectance spectra method. SHEIN et al [9] investigated the electronic band structure, density of states and inter-atomic bonding picture for BiOCuS. The results showed that the BiOCuS phase behaves as an ionic semiconductor with the calculated indirect band gap of about 0.48 eV. Very recently, superconductivity below TC=5.8 K was reported for the tetragonal Cu-deficient BiOCu1-xS samples, which declared BiOCuS as a parent phase for novel layered superconductors [11-13].
To the best of our knowledge, there are no reports on the elastic properties of BiOCuS. However, it is important for fundamental physics and potential applications to study elastic properties of BiOCuS. Elastic properties provide information on interatomic potentials, interatomic bonding, equations of state, phonon spectra, specific heat capacity, thermal expansion, Debye temperature, etc [14]. In addition, elastic properties are essential for many practical applications related to the mechanical properties of solid: load deflection, internal strain, thermoelastic stress, sound velocities and fracture toughness [14].
In this study, the electronic structures, chemical bonding and elastic properties of the tetragonal phase of BiOCuS were studied using first-principles calculations based on DFT.
2 Calculation details
The first principles calculations described here were based on DFT using a plan-wave expansion of the wave function [15,16]. The exchange correlation energy was calculated by the GGA with the Perdew-Burke- Ernzerhof (PBE) function [17]. The ionic cores were represented by ultra-soft pseudopotentials for Bi, O, Cu and S atoms. The Bi 6s26p3 electrons, O 2s22p4 electrons, Cu 3d104s1 electrons, and S 3s23p4 electrons were explicitly treated as valence electrons. The Monkhorst and Pack scheme of k-point sampling was used for integration over the first Brillouin zone [18]. The cutoff energy was chose to be 600 eV, and the Brillouin-zone sampling k-point set mesh parameters are 10×10×5. This set of parameters assured the total energy convergence of 5.0×10-6 eV/atom, the maximum force of 0.01 eV/?, the maximum stress of 0.02 GPa and the maximum displacement of 5.0×10-4 ?.
3 Results and discussion
3.1 Geometry and structure properties
The crystal structure of the tetragonal phase of BiOCuS belongs to the space group P4/nmm, Z=2 (ZrCuSiAs type), where blocks [BiO] are sandwiched with [CuS] blocks as depicted in Fig. 1. There are four inequivalent atomic positions: Bi at 2c site (1/4, 1/4, zBi), O at 2a site (3/4, 1/4, 0), Cu at 2b site (3/4, 1/4, 1/2), and S at 2c site (1/4, 1/4, zS) [7], where zBi and zS are the internal coordinates of Bi and S, respectively. The experimental lattice parameters are a=b=3.8691 ? and c=8.5602 ?, and the internal coordinates of Bi and S, were reported as zBi=0.14829 and zS=0.6710 [7], respectively.
At the first stage, the full structural optimization of this phase was performed both over the lattice parameters and the atomic positions including the internal coordinate zBi and zS. The calculated optimization lattice parameters a, c, V and atomic coordinates compared with available experimental data [7] for BiOCuS are summarized in Table 1, which shows that the calculated values of GGA calculation are in agreement with the experimental results. The differences between our values and the experimental data may be due to the use of an approximate DFT. It is well known that the GGA leads to volume slightly overestimated in relation to the experimental value.

Fig. 1 Crystal structure of BiOCuS
Table 1 Calculated lattice parameters and atomic internal coordinate compared with available experimental data [7] for BiOCuS

3.2 Electronic and chemical bonding
The calculated energy band structure of BiOCuS along with the high-symmetry points of the Brillouin zone by GGA is shown in Fig. 2. The top of the valence band is taken as the zero of energy. This compound is found to have indirect band gap. The valence band maximum (VBM) is at the Z point and the conduction band minimum (CBM) is on the M-Γ line. The calculated band gap value is 0.503 eV by GGA, which is smaller than the experimental value of 1.1 eV [7] due to the well-known underestimation of conduction band state energies in DFT calculations.
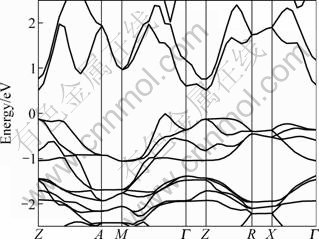
Fig. 2 GGA calculated band structure of BiOCuS along some high-symmetry lines in Brillouin zone
The total and partial densities of states (DOS) of BiOCuS calculated by GGA are shown in Fig. 3 and Fig. 4. It is found that the lower valence bands (between -20.56 and -17.67 eV) are essentially dominated by O-2s states and show hybridization with Bi-6s and Bi-6p states. The structure situated in the range from -14.46 to -12.30 eV originates predominantly from S-3s, and the one in the range from -12.10 to -8.73 eV originates predominantly from Bi-6s states. The upper valence bands are composed mainly of Cu-3d character and show hybridization with S-3p state. The conduction bands are dominated by Bi-6p state and hybridized Cu-4p and S-3p states. The calculated results are in agreement with the previous results [9].
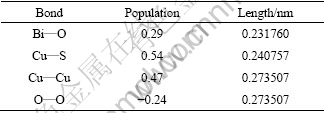
The Mulliken bond population is useful for evaluating the bonding character in a material. A high value of the bond population indicates a covalent bond, and a low value indicates an ionic bond. Positive and negative values indicate bonding and anti-bonding states, respectively [19,20]. The Mulliken atomic population of BiOCuS reported in Table 2 shows a significant charge transfer from [BiO] block to [CuS] blocks, indicating that the internal of [BiO] block and the [CuS] block is ionic character. The bond population reported in Table 3 shows that the intra-block of Bi-O and Cu-S is covalent character. The chemical bonding in BiOCuS has predominantly ionic character with mixed covalent-ionic character, which is in agreement with the results of Ref. [12].
3.3 Elastic properties
Elastic properties are very important for materials because they provide information on the interatomic potentials and relate to various fundamental solid state phenomena, such as interatomic bonding, equations of state, phonon spectra as well as specific heat capacity, thermal expansion, Debye temperature [21,22].
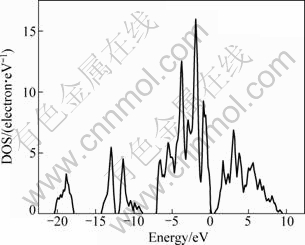
Fig. 3 Total DOS of BiOCuS (the zero of energy is at the Fermi level)
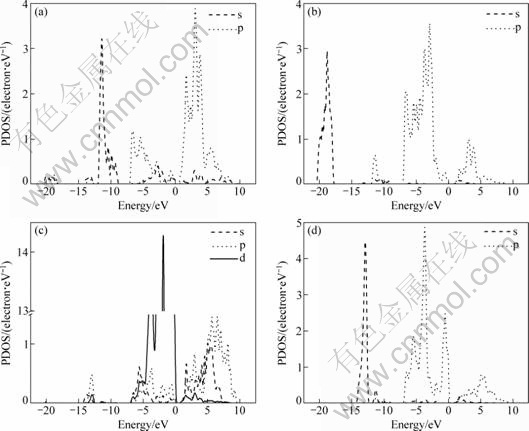
Fig. 4 PDOS of Bi (a), O (b), Cu (c) and S(d) of BiOCuS calculated by GGA
Table 2 Mulliken atomic population of BiOCuS

Table 3 Mulliken bond population of BiOCuS
Elastic constants are defined by means of a Taylor expansion of the total energy, namely the derivative of the energy as a function of a lattice strain [15,16]. The tetragonal phase BiOCuS crystal has six independent single crystal elastic constants C11, C33, C44, C66, C12 and C13 [23]. The GGA calculated Cij are presented in Table 4. For the tetragonal crystal, its mechanical stability requires that its independent elastic constants should satisfy the Born’s stability criteria [23]:
 (1)
(1)
Table 4 Calculated single crystal elastic constants Cij, bulk modulus and compressibility coefficient β of BiOCuS

From Table 4, we can see that these criteria are all satisfied, which indicates that BiOCuS is mechanically stable. The single crystal bulk modulus B of BiOCuS is about 85 GPa, which is smaller than that of the ZrCuSiAs-type Fe-based superconductor parent compound LaFeAsO (98 GPa) and is close to that of YZnAsO/LaZnAsO (78/87 GPa), meaning that BiOCuS is relatively soft material [23-25]. The polycrystal bulk modulus, B, and shear modulus, G are estimated using the Voigt-Reuss-Hill approach in the following forms [23-25]:
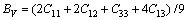 (2)
(2)
 (3)
(3)
 (4)
(4)
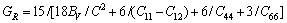 (5)
(5)
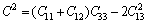 (6)
(6)
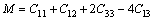 (7)
(7)
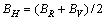 (8)
(8)
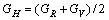 (9)
(9)
Elastic modulus E and Poisson ratio v are estimated by
 (10)
(10)
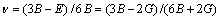 (11)
(11)
All the calculated results are presented in Table 5. It can be seen that the value of B/G ratio for BiOCuS is 2.16, which is larger than the critical value (1.75) separating ductile and brittle materials [23-28], indicating that BiOCuS behaves in a ductile manner. For LaOFeAs and LaOFeP, the values of B/G ratio are 1.74 and 1.42, respectively [23,24]. From the point of application, BiOCuS is more readily machinable.
Table 5 Calculated polycrystalline elastic constants, elastic modulus E, Poisson ratio ν and B/G of BiOCuS

It is known that the values of the Poisson ratio (ν) are minimal for covalent materials (ν=0.1), and increase for ionic systems [29]. In our case, the calculated Poisson ratio is 0.2992, which means a sizable ionic contribution in intra-bonding.
3.4 Debye temperature
The Debye temperature (ΘD) is a fundamental parameter of a material, which is linked to many physical properties, such as specific heat capacity, elastic constants, and melting point. At low temperatures, the vibration excitations arise solely from acoustic vibrations. Hence, at low temperatures, the Debye temperature calculated from elastic constants is the same as that determined from specific heat capacity measurements. It can be obtained from the average wave velocity by the following equation [29]:
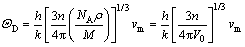 (12)
(12)
where h is the Planck constant; k the Boltzmann constant; NA the Avogadro number; ρ the density; n the number of atoms per formula unit; M the relative molecular mass per formula unit; V0 the volume of per formula unit; vm is the average wave velocity and is approximately estimated by
 (13)
(13)
where vs and vl are transverse and longitudinal elastic wave velocities, respectively, which can be determined by the shear modulus G and the bulk modulus B from Navier’s equation:
 ,
,  (14)
(14)
The calculated average, longitudinal and transverse elastic wave velocities and Debye temperature for BiOCuS are given in Table 6. However, no other theoretical or experimental data exist for comparison with the present values.
Table 6 Calculated Debye temperature ΘD of BiOCuS

4 Conclusions
1) The GGA calculated structural parameters of the tetragonal phase BiOCuS are in agreement with the experimental data.
2) The electronic band structures present that BiOCuS has indirect band gap with 0.503 eV by GGA. The DOS and the PDOS calculations show that the DOS near the Fermi level is mainly from the Cu-3d state.
3) The chemical bonding was analyzed, which shows that BiOCuS has mainly ionic character with mixed covalent-ionic character.
4) The elastic constants were calculated and the bulks and shear modulus, elastic modulus, Poisson ratio were derived. All results show that BiOCuS is mechanically stable and behaves in a ductile manner.
References
[1] Kamihara Y, Hiramatsu H, Hirano M, Kawamura R, Yanagi H, Kamiya T, Hosono H. Iron-based layered superconductor: LaOFeP [J]. J Am Chem Soc, 2006, 128: 10012-10014.
[2] Watanabe T, Yanagi H, Kamiya T, Kamihara Y, Hiramatsu H, Hirano M, Hosono H. Nickel-based oxyphosphide superconductor with a layered crystal structure, LaNiOP [J]. Inorganic Chemistry, 2007, 46(19): 7719-1123.
[3] Kamihara Y, Watanabe T, Hirano M, Hosono H. Iron-based layered superconductor La[O1-xFx]FeAs (x=0.05-0.12) with Tc=26 K [J]. J Am Chem Soc, 2008, 130: 3296-3297.
[4] Wen H H. Developments and perspectives of iron-based high-temperature superconductors [J]. Adv Mater, 2008, 20(19): 3764-3775.
[5] Ren Z A, Zhao Z X. Research and prospects of iron-based superconductors [J]. Adv Mater, 2009, 21(45): S4584-S4583.
[6] Poettgen R, Johrendt D. Materials with ZrCuSiAs type structure [J]. Z Naturforsch, 2008, 63b: 1135-1148.
[7] Hiramatsu H, Yanagi H, Kamiya T, Ueda K, Hirano M, Hosono H. Crystal structures, optoelectronic properties, and electronic structures of layered oxychalcogenides MCuOCh (M=Bi, La; Ch=S, Se, Te): Effects of electronic configurations of M3+ ions [J]. Chem Mater, 2008, 20: 326-334.
[8] Sheets W C, Stampler E S, Kabbour H, Bertoni M I, Laurent ario C, Mason T O, Marks T J, Poeppelmeier K R. Facile synthesis of BiCuOS by hydrothermal methods [J]. Inorg Chem, 2007, 46: 10741-10748.
[9] Shein I R, Ivanovskii A L. Electronic band structure and inter-atomic bonding in tetragonal BiOCuS as a parent phase for novel layered superconductors [J]. Solid State Commun, 2010, 150: 640-643.
[10] Kusainova A M, Berdonosov P S, Akselrud L G, Kholodkovskaya L N, Dolgikh V A, Popovkin B A. New layered compounds with the general composition (MO)(CuSe), where M=Bi, Nd, Gd, Dy, and BiOCuS: Syntheses and crystal structure [J]. J Solid State Chem, 1994, 112: 189-191.
[11] Ubaldini A, Giannini E, Senatore C, Marel D. BiOCuS: A new superconducting compound with oxypnictide-related structure [J]. Physica C, 2010, 470: S356-S357.
[12] Mazin I I. Superconductivity and magnetism in CuBiSO from first principles [J]. Phys Rev B, 2010, 81: R140508
[13] Pal A, Kishan H, Awana V P S. Synthesis and structural details of BiOCu1-xS: possible new entrant in a series of exotic superconductors [J]. J Supercond Nov Magn, 2010, 23: 301-304.
[14] Peng F, Chen D, Yang X D. First-principles calculations on elasticity of OsN2 under pressure [J]. Solid State Commun, 2009, 149: 2135-2138.
[15] Segall M D, Lindan Philip J D, Probert M J, Pickard C J, Hasnip P J, Clark S J, Payne M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[16] Clark S J, Segall M D, Pickard C J, Hasnip P J, Probert M J, Refson K, Payne M C. First principles methods using CASTEP [J]. Zeitschrift fuer Kristallographie, 2005, 220(5-6): 567-570.
[17] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple [J]. Phys Rev Lett, 1996, 77: 3865-3868.
[18] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations [J]. Phys Rev B, 1976, 13: 5188-5192.
[19] Hermet P, Goumri-Said S, Kanoun M B, Henrard L. First-principles investigation of the physicalproperties of magnesium nitridoboride [J]. J Phys Chem C, 2009, 113: 4997-5003.
[20] Xia Q L, Yi J H, Li Y F, Peng Y D, Wang H Z, Zhou C S. First-principles investigations of the band structure and optical properties of γ-boron [J]. Solid State Commun, 2010, 150: 605-608.
[21] Ponce C A, Casali R A, Caravaca M A. Ab initio study of mechanical and thermo-acoustic properties of tough ceramics: Applications to HfO2 in its cubic and orthorhombic phase [J]. J Phys Condens Mater, 2008, 20: 045213.
[22] Bouhemadou A, Khenata R, Chegaar M, Maabed S. First-principles calculations of structural, elastic, electronic and optical properties of the antiperovskite AsNMg3 [J]. Phys Lett A, 2007, 371: 337-343.
[23] Shein I R, Ivanovskii A L. Elastic properties and chemical bonding in ternary arsenide SrFe2As2 and quaternary oxyarsenide LaFeAsO─Basic phases for new 38-55K superconductors from first principles [J]. Physica C, 2009, 469: 15-19.
[24] SHEIN I R, IVANOVSKII A L. Elastic properties of quaternary oxyarsenide LaOFeAs and LaOFeP as basic phases for new 26-52 K superconducting materials from first principles [J]. Scripta Materialia, 2008, 59: 1099-1102.
[25] SHI Yi-ming, YE Shao-long. Chemical bonding and elastic properties of quaternary arsenide oxides YZnAsO and LaZnAsO investigated by first principles [J]. Transactions of Nonferrous Metals Society of China, 2011, 21: 1378-1382.
[26] HILL R. The elastic behaviour of a crystalline aggregate [J]. Proc Phys Soc London A, 1952, 65: 349-355.
[27] Xia Qing-lin, Yi Jian-hong, Peng Yuan-dong, Wang Hong-zhong, Zhou Cheng-shang. First-principles study of electronic structure and elastic properties of C doping Mg(B1-xCx)2 [J]. Materials Science and Engineering of Powder Metallurgy, 2011, 16(1): 7-12. (in Chinese)
[28] Haines J, Leger J M, Bocquillon G. Sythesis and design of superhard materials [J]. Annu Rev Mater Res, 2001, 31: 1-23.
[29] Anderson O L. A simplified method for calculating the Debye temperature from elastic constants [J]. J Phys Chem Solids, 1963, 24(7): 909-917.
BiOCuS电子结构、化学键和弹性性质的第一性原理研究
潘留仙1,夏庆林2,叶绍龙3,丁 宁1,刘自然1
1. 湖南涉外经济学院 电气与信息工程学院,长沙 410205;
2. 中南大学 物理与电子学院,长沙 410083;
3. 中南大学 冶金科学与工程学院,长沙 410083
摘 要:利用基于密度泛函理论(DFT)的广义梯度近似(GGA)研究四方相BiOCuS的电子结构、化学键和弹性性质。能带结构显示,BiOCuS为间接带隙半导体,带隙宽为0.503 eV;态密度和分态密度的结果表明,费米能级附近的态密度主要来自Cu-3d态。布居分析表明,BiOCuS中的化学键具有以离子性为主的混合离子-共价特征。计算得到四方相BiOCuS的晶格参数、体模量、剪切模量和单晶的弹性常数,由此导出弹性模量和泊松比。结果表明,BiOCuS是力学稳定的,且具有一定的延展性。
关键词:BiOCuS;第一性原理;电子结构;化学键;弹性性质
(Edited by YUAN Sai-qian)
Foundation item: Project (60571043) supported by the National Natural Science Foundation of China; Project (11JJ2002) supported by the Natural Science Foundation of Hunan Province, China
Corresponding author: PAN Liu-xian; +86-731-88127220; E-mail: PLX123@126.com; XIA Qing-lin; +86-731-88836692; E-mail: qlxia@csu.edu.cn
DOI: 10.1016/S1003-6326(11)61305-8

