
Crystal structure of Mg3Pd from first-principles calculations
DENG Yong-he(邓永和)1, WANG Tao-fen(王桃芬)1, ZHANG Wei-bing(张卫兵)1,
TANG Bi-yu(唐璧玉)1, ZENG Xiao-qin(曾小勤)2, DING Wen-jiang(丁文江)2
1. Key Laboratory of Low Dimensional Materials & Application Technology of Chinese Ministry of Education, Xiangtan University, Xiangtan 411105, China;
2. Light Alloy Net Forming National Engineering Research Center, School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200030, China
Received 8 May 2007; accepted 21 September 2007
Abstract: Crystal structure of Mg3Pd alloy was studied by first-principles calculations based on the density functional theory. The total energy, formation heat and cohesive energy of the two types of Mg3Pd were calculated to assess the stability and the preferentiality. The results show that Mg3Pd alloy with Cu3P structure is more stable than Na3As structure, and Mg3Pd alloy is preferential to Cu3P structure. The obtained densities of states and charge density distribution for the two types of crystal structure were analyzed and discussed in combination with experimental findings for further discussion of the Mg3Pd structure.
Key words: crystal structure; Mg3Pd; first-principles; formation heat; cohesive energy; density of states(DOS); charge distribution
1 Introduction
During the past decades, the global greenhouse warming caused mainly by CO2 emission is becoming more and more worrying. It is now well accepted that hydrogen is an ideal non-polluting vector of energy for the future. This means that we would go from a “petroleum economy” to a “hydrogen economy”. The widespread of hydrogen requires some progresses especially in the field of storage and mass production. Hydrogen-storage materials have attracted much attention in view of the development of hydrogen energy systems. Metal hydrides appear as a suitable route for storage compared with pressurized or liquefied mode. Among all materials, Mg is one metal hydrogen-storage material that can reach the highest mass capacity (i.e. 7.6%). According to its abundance on the earth’s crust and its low cost, it is often considered as the most promising material. However, the absorption and desorption of hydrogen molecule take place at high temperature and have very low-efficiency and low-rate. To solve these problem and increase its storage capacity, a considerable number of work aimed at improving the microstructure and researching for new hydrides or alloys has been reported[1-2]. HIGUCHI et al[3] and CHECCHETTO et al[4] investigated some advantageous hydrogen storage properties of magnesium-palladium intermetallic compounds or alloys. GAOTO et al[2] reported X-ray diffraction patterns of Mg-x%Pd(mole fraction) samples (x=10, 18, 22, 26, 33, 38, 50, 55) prepared by 4 h exposure to Ar gas at 1 073 K and 2 GPa, and their hydrogen storage properties. However, they did not give the structure information such as crystal lattice parameters of Mg-Pd systems except Mg4Pd. In the recent study, FERRO[5], RANGE and HAFFNER[6] investigated the crystal structure of Mg3Pd based on different crystal structure models, respectively. However, they still did not determine the structural stability and preferentiality.
The crystal structure with the first-principles plane-wave pseudopotential method was investigated based on the density functional theory(DFT). The total energies, formation heats, cohesive energies, electron density of states and charge distributions were studied and discussed for determination of the crystal structure of Mg3Pd.
2 Model and method of calculation
The intermetallic compound of Mg3Pd was firstly studied by FERRO[5] who assigned the Na3As-type structure (Pearson symbol: hP8, space group: P63/mmc, number: 194, Z=2). However, the recent investigation of range showed that the Mg3Pd was very likely to adopt the Cu3P-type structure (Pearson symbol: hp24, space group:  number: 165, Z=6)[6-7]. MOKONGO et al[8] thought that Cu3P-type structure can be derived from the Na3As-type structure by a slight deformation of the CN 11 polyhedra and the lattice constants follow the relationship
number: 165, Z=6)[6-7]. MOKONGO et al[8] thought that Cu3P-type structure can be derived from the Na3As-type structure by a slight deformation of the CN 11 polyhedra and the lattice constants follow the relationship 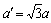 and c′=c (a′=7.987 nm and c′=8.422 nm). In this work, we will provide further structural information of the Mg3Pd intermetallic mainly based on the Cu3P-type and Na3As-type structural model.
and c′=c (a′=7.987 nm and c′=8.422 nm). In this work, we will provide further structural information of the Mg3Pd intermetallic mainly based on the Cu3P-type and Na3As-type structural model.
The present work was based on density functional theory(DFT) at the level of the generalized gradient approximation(GGA) (using the PW-91 exchange- correlation functional). The method was implemented in the Vienna ab-initio simulation package(VASP) program. Ultrasoft pseudopotential was used to describe the election-ion interaction, and the energy cutoff(Ecut) of atomic wave functions was set at 340 eV, and the 4×4×4 and 6×6×6 k point meshes of the Monkhorst- Pack(MP) algorithm were used to sample the super cell of Cu3P-type and Na3As-type Mg3Pd bulk respectively. These parameters are shown to give well-converged total-energies to less than 1.0 meV/atom (≈0.1 kJ/mol) [9]. Ionic relaxation and electronic energy minimization were performed using the conjugate gradient (CG) algorithm. Density of states(DOS) and charge distri- butions were calculated with finer k point meshes, and the tetrahedron method with Bloch corrections was used.
3 Results and discussion
3.1 Lattice parameters and electronic total energy
The lattice parameters of hcp Mg, fcc Pd, Mg3Pd of Cu3P-type and Na3As-type structure were firstly calculated from the minimized total energy, and the obtained results are listed in Table 1. It can be found that the present lattice parameters a and c/a of hcp Mg are 0.319 5 nm and 1.624 1, which are in good agreement with the experimental values of 0.321 nm and 1.623[10] and other GGA calculations[11]. The lattice parameter a of fcc Pd is 0.395 1 nm, which is also in good agreement with the experimental value of 0.389 nm[12] and other GGA calculations[13], showing good accuracy of the present calculations. The present first-principles calculations show that the electronic total energy of Na3As-type (there are 6 magnesium atoms and 2 atoms palladium atoms in the super cell) and Cu3P-type (there are 18 magnesium atoms and 6 atoms palladium atoms in the super cell) structure Mg3Pd are -69.390 0 eV and -22.956 6 eV, respectively. This results indicate that the Cu3P-type structure is more stable than Na3As-type structure, which is accordance with the suggestion of MAKONGO et al[8]. The calculated Mg3Pd lattice parameters (a and c/a) of Cu3P-type and Na3As-type structure are 0.802 6 nm and 1.048 8, 0.465 1 nm and 1.805 7, respectively. It can also be found the Cu3P-type and Na3As-type structure lattice parameters have the following relationship, and c′≈c, which is in line with Ref.[8]. Based on the slight difference of the electronic total energy and the relationship between the lattice parameters, Cu3P-type crystal structure is likely to derive from three same Na3As-type crystal structure cells through a slight deformation.
Table 1 Contrast of VASP calculation and experiments or other GGA calculation

3.2 Formation heat and cohesive energy
In order to assess the thermodynamic aspect of the two crystal structure types of Mg3Pd, the formation heat is an important parameter. At the same time, the formation heat could also explain and deepen the understanding of the structural stability of alloys system[14]. The formation heat of Mg3Pd alloy per atom can be calculated by[15]
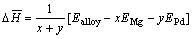 (2)
(2)
where  is the average formation heat of Mg3Pd alloy per atom; Ealloy is the electronic total energy of primitive cell of Mg3Pd; EMg and EPd are the total energy of single magnesium and palladium atom at solid state, respectively; x and y are the number of magnesium and palladium atom in primitive cell of Mg-Pd alloy. The average formation heat of Mg3Pd per atom for Na3As-type and Cu3P-type is expressed as
is the average formation heat of Mg3Pd alloy per atom; Ealloy is the electronic total energy of primitive cell of Mg3Pd; EMg and EPd are the total energy of single magnesium and palladium atom at solid state, respectively; x and y are the number of magnesium and palladium atom in primitive cell of Mg-Pd alloy. The average formation heat of Mg3Pd per atom for Na3As-type and Cu3P-type is expressed as  and
and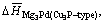 respectively.
respectively.
The obtained formation heats of Mg3Pd per atom for Cu3P-type and Na3As-type are -0.440 1 eV and -0.418 4 eV, respectively. It is clearly shown that 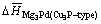 is lower than
is lower than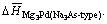 Hence, the alloying ability of Mg3Pd for Cu3P-type crystal structure is thermodynamically stronger.
Hence, the alloying ability of Mg3Pd for Cu3P-type crystal structure is thermodynamically stronger.
The stability of crystal structure is correlated to its cohesive energy[16], and the cohesive energy is often defined as energy needed when crystal is decomposed into the single atom. Hence, the lower the cohesive energy is, the more stable the crystal structure is[16]. In this work, average cohesive energy per atom for Mg-Pd binary alloy family was calculated using the following expression[15]:
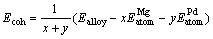 (2)
(2)
where Ealloy is the electronic total energy of primitive cell of Mg3Pd, x and y also are the number of magnesium and palladium atoms in primitive cell of Mg-Pd alloy;  and
and  are the electronic total energies of single Mg atom and Pd atom in freedom states. They are -0.045 8 eV and -1.478 4 eV for Mg and Pd free atoms, respectively. The obtained average cohesive energies of Mg3Pd per atom for Cu3P-type and Na3As-type are -2.487 3 eV and -2.465 6 eV, respectively. It can be clearly found that the average cohesive energy of Cu3P-type is also lower than that of Na3As-type. Hence, Mg3Pd of the Cu3P-type crystal structure is slightly more stable than that of Na3As-type.
are the electronic total energies of single Mg atom and Pd atom in freedom states. They are -0.045 8 eV and -1.478 4 eV for Mg and Pd free atoms, respectively. The obtained average cohesive energies of Mg3Pd per atom for Cu3P-type and Na3As-type are -2.487 3 eV and -2.465 6 eV, respectively. It can be clearly found that the average cohesive energy of Cu3P-type is also lower than that of Na3As-type. Hence, Mg3Pd of the Cu3P-type crystal structure is slightly more stable than that of Na3As-type.
From the above calculation results of formation heats and cohesive energies, it could be expected that the Cu3P-type crystal is overall preferential to Na3As-type.
3.3 Density of states(DOS)
For further understanding of the structural stability and microcosmic mechanism of the Mg3Pd alloy, total and partial density of states(DOS) of Mg3Pd for Cu3P-type and Na3As-type crystal structure are performed, and the results are shown in Fig.1. For comparison and analysis, the total and partial density of states(DOS) of hcp-Mg and fcc-Pd are also calculated, as shown in Fig.1. The partial DOS of pure hcp-Mg is much smaller than total density of states, and the s and p partial DOS of pure fcc-Pd crystal is also much less than d partial density of states. And the partial density of states of Mg(s), Mg(P), Pd(s) and Pd(p) of two types of Mg3Pd is also more less than their total density of states and the Pd (d) partial density of states.

Fig.1 Total and partial density of states of hcp-Mg, fcc-Pd and Mg3Pd for Cu3P -type and Na3As -type
From the total DOS in Fig.1, it can be seen that the main bonding peaks of both Mg3Pd structure are in the energy range between about -3.4 and -2.6 eV under Fermi level, which are mainly dominated by the valence electron numbers of Pd(d) and Mg(p) orbits and also affected by part of Mg(s) and Pd(p) orbits. In this way, the dominant bonding possesses the characteristic of significant p-d hybridization, which leads to the relatively high formation heat and cohesive energy of Mg3Pd. This feature is also found in other alloy of 3d transition metal and magnesium[17]. It should be noted that due to interaction of Mg and Pd in the Mg3Pd, there are large variation in the valence electrons of Pd(d) orbits compared with partial density of states of fcc-Pd. The partial DOS of Pd(d) in the fcc-Pd reveals that the main bonding peaks are in the energy range between about -5.0 eV under Fermi level and 0.5 eV upon Fermi level. However the partial DOS of Pd(d) in the Mg3Pd reveals that the main bonding peaks are in the energy range between about -3.4 eV and -2.6 eV under Fermi level. This shows that in the alloy, low energy end of Pd(d) DOS shifts up while high energy end of Pd(d) DOS shifts down, the width of main peak region is narrower and the density of states increases apparently, showing very strong interaction between Pd and Mg.
The electronic structures of two type crystal structures also can be further compared and analyzed as follows. Because the atom number of Mg3Pd for Cu3P-type is triple of that for Na3As-type, the electronic total and partial densities of states of Mg3Pd for Cu3P- type are also triple of those for Na3As-type. From the total and partial densities of states for two type of crystal structures, the difference of the electronic structures can be further perceptible although it is very small. As shown in Fig.1, although two high peaks for both Mg3Pd structure appear between -2.7 eV and -3.0 eV respectively, the two energy peaks (about -2.7 eV and -3.0 eV) in Na3As-type structure is higher and sharper, showing that electron number of Na3As-type is more than Cu3P-type around the two energy positions. The other peaks of Cu3P-type from about -3.4 eV to -2.6 eV under Fermi level are clearly higher than those of Na3As-type, implying that the electron number of Cu3P-type is more uniform in this energy region. This shows that density of states of Cu3P-type is more helpful for alloy formation.
Further analysis reveals that in the energy range between -10 and 5 eV, the bonding electron numbers per atom for Cu3P-type and Na3As-type are 5.179 8 and 5.176 1, respectively. Because the electrons in low- energy region of Fermi level are the mainly bonding electron, when more electrons are in low-energy region, the bonding electron increases, and the interaction between valence electrons of crystal is intensified, so the stability of crystal structure is better[18]. From the density of states, it has a very good agreement with the formation heat and cohesive energy, hence, it could be also expected that the Cu3P-type crystal is preferential to Na3As-type.
3.4 Charge density
In order to further understand the microscopic mechanism of bonding in Mg3Pd alloy, charge density distributions were investigated. The charge distributions calculated on (010) plane for Cu3P-type and (100) plane for Na3As-type are shown in Fig.2(a) and Fig.2(b), respectively. It is clear that the feature of covalent bond between Mg and Pd atoms exhibits apparently, and charge distributions between Mg and Pd atoms for two types of crystal structure are very similar. Nevertheless, according to the DOS and bonding electron number mentioned above, the charge density overlapping between Mg atom and Pd atom in the Cu3P-type Mg3Pd crystal should be enhanced very slightly compared with that in the Na3As-type structure. So, the bond strength between Mg and Pd atoms Cu3P-type structure should also be increased very slightly.
According the above discussion and analysis of the charge density distributions, it could be also expected that the Cu3P-type crystal is preferential to Na3As-type.
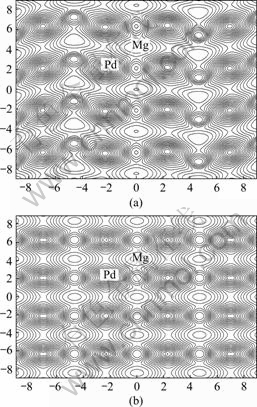
Fig.2 Charge distribution of Mg3Pd on (100) plane for Cu3P- type (a) and (010) plane for Na3As-type (b)
4 Conclusions
1) The calculation results show that Cu3P-type crystal structure is more stable, and the alloying ability of Cu3P-type Mg3Pd is also stronger.
2) The comparison and analysis of total and partial density of states(DOS) and charge distributions show that Cu3P-type is more helpful for alloy formation. So Mg3Pd alloy is overall preferential to Cu3P-type structure.
References
[1] ORIMO S, FUJII H, IKEDA K. Notable hydriding properties of a nanostructured composite material of the Mg2Ni-H system synthesized by reactive mechanical grinding [J]. Acta Mater, 1997, 45: 331-341.
[2] GAOTO Y, KAKUTA H, KAMEGAWA A, TAKAMURA H, OKADA M. High-pressure synthesis of novel hydride in Mg-M systems (M=Li, Pd) [J]. J Alloys Compd, 2005, 404/406: 448-452.
[3] HIGUCHI K, YAMAMOTO K, KAJIOKA H, TOIYAMA K, HONDA M, ORIMO S, FUJII H. Hydrogen storage in magnesium clusters: Quantum chemical study [J]. J Alloys Compd, 2002, 330: 526-530.
[4] CHECCHETTO R, BRUSA R S, BAZZANELLA N, KARWASZ G P, SPAGOLLA M, MIOTELLO A, MENGUCCI P, DI CRISTOFORO A. Structural evolution of nanocrystalline Pd-Mg bilayers under deuterium absorption and desorption cycles [J]. Thin Solid Film, 2004, 469/470: 350-355.
[5] FERRO R. Research on the alloys of noble metals with the more electro-positive elements: Micro graphic and rentgeno graphic examination of the magnesium-palladium alloys [J]. J Less-Common Met, 1959, 1: 424-438.
[6] RANGE K J, HAFFNER P. Structure refinement of AuMg3, IrMg3 and IrMg2.8 [J]. J Alloys Compd, 1993, 191: L5-L7.
[7] RANGE K J, HAFFNER P. A redetermination of the crystal structure of trimagnesium platinum, Mg3Pt [J]. J Alloys Compd, 1991, 183: 430-437.
[8] MAKONGO J P A, KUDLA C, PROTS Y, NIEWA R, BURKHARDT U, KREINER G. Crystal structure of trimagnesium monopalladium, Mg3Pd [J]. Z Kristallogr. NCS, 2005, 220: 289-290.
[9] ZHANG Wei-bing, HU Yu-lin, HAN Ke-li, TANG Bi-yu. Pressure dependence of exchange interactions in NiO [J]. Phys Rev B, 2006, 74: 054421-054425.
[10] KITTEL C. Introduction to solid state physics [M]. 7th ed. New York: Wiley, 1996.
[11] SCHR?DER E, FASEL R, KIEJNA A. O adsorption and incipient oxidation of the Mg(0001) surface [J]. Phys Rev B, 2004, 69: 115431-115438.
[12] MASSARD R, UZIO D, THOMAZEAU C, PICHON C, ROUSSET J L, BERTOLINI J C. Strained Pd overlayers on Ni nanoparticles supported on alumina and catalytic activity for buta-1,3-dience selective hydrogenation [J]. Journal of Catalysis, 2007, 245: 133-143.
[13] ALFONSO D R. Initial incorporation of sulfur into the Pd(111) surface: A theoretical study [J]. Surface Science, 2006, 600: 4508-4516.
[14] SONG Y, GUO Z X, YANG R. Influence of selected alloying elements on the stability of magnesium dihydride for hydrogen storage applications: A first-principle investigation [J]. Phys Rev B, 2004, 69: 094205-094215.
[15] SAHU B R, Mater. Electronic structure and bonding of ultralight LiMg [J]. Sci Eng B, 1997, 49: 74-78.
[16] ZUBOV V I, TRETIAKOV N P, TEIXEIRA RABELO J N, SANCHEZ ORTIZ J F. Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a Van der Waals crystal - fullerene C60 [J]. Phys Lett A, 1995, 198(1): 223-227.
[17] ZHOU Dian-wu, PENG Ping, LIU Jin-shui, CHEN lu, HU Yan-jun. First-principles study on structural stability of 3d transition metal alloying magnesium hydride [J]. Trans Nonferrous Met Soc China, 2006, 16(1): 23-32.
[18] GHOSH G. First-principles calculations of structural energetics of Cu-TM (TM=Ti, Zr, Hf) intermetallics [J]. Acta Materialia, 2007, 55: 3347-3374.
Foundation item: Project(07A070) supported by the Key Program of Educational Department of Hunan Province, China; Project(KF0504) supported by the Open Project Program of the Key Laboratory of Low Dimensional Materials & Application Technology, Ministry of Education, China
Corresponding author: TANG Bi-yu; Tel: +86-13187321190; E-mail: tangbiyu@xtu.edu.cn
(Edited by LI Xiang-qun)

