
First-principles study on structural stability of 3d transition metal alloying magnesium hydride
ZHOU Dian-wu(�ܵ���), PENG Ping(�� ƽ), LIU Jin-shui(����ˮ), CHEN L��(�� ��), HU Yan-jun(����)
School of Materials Science and Engineering, Hunan University, Changsha 410082, China
Received 13 June 2005; accepted 14 September 2005
Abstract: A first-principles plane-wave pseudopotential method based on the density functional theory was used to investigate the energy and electronic structure of magnesium hydride (MgH2) alloyed by 3d transition metal elements. Through calculations of the negative heat formation of magnesium hydride alloyed by X (X denotes 3d transition metal) element, it is found that when a little X (not including Sc) dissolves into magnesium hydride, the structural stability of alloying systems decreases, which indicates that the dehydrogenation properties of MgH2 can be improved. After comparing the densities of states(DOS) and the charge distribution of MgH2 with or without X alloying, it is found that the improvement for the dehydrogenation properties of MgH2 alloyed by X attributes to the fact that the weakened bonding between magnesium and hydrogen is caused by the stronger interactions between X (not including Cu) and hydrogen. The calculation results of the improvement for the dehydrogenation properties of MgH2��X (X=Ti, V, Mn, Fe, Co, Ni, Cu) systems are in agreement with the experimental results. Hence, the dehydrogenation properties of MgH2 are expected to be improved by addition of Cr, Zn alloying elements.
Key words: magnesium hydride; plane-wave pseudopotential theory; heat formation; electronic structure
1 Introduction
Nowadays, many efforts have been spent to develop hydrogen storage materials for non-polluting applications. Magnesium hydrogen systems can act as a thermal energy storage system in the future. Magnesium-based hydrogen storage alloys have been extensively researched due to their high hydrogen storage capacity, light mass and low cost. However, the slow hydriding and dehydrogenating kinetics and high dissociation temperature caused by its relatively high stability limit its practical application for hydrogen storage. The main objective of many current studies has been done to reduce the high Mg-H binding energy by alloying additions, so as to reduce the sorption temperature. Numerous studies have been carried out in order to identify a suitable alloy that absorbs hydrogen close to room temperature and desorbs hydrogen at a temperature low enough to use the waste heat of exhaust gas. Both nanocrystallization and addition of transition metal catalysts demonstrated to improve the hydrogen sorption kinetics of Mg. Recent experiment investigations[1��4] have shown that the mechanical alloying of MgH2 and 3d transition metal elements, for example, Ni, Co, Mn, Cu, Ti, Fe, V, effectively improves the hydriding and dehydrogenating kinetics of MgH2 at high temperature. As far as 3d transition metal is concerned, Ti and V, when mixed with the magnesium hydrides by ball milling, are better catalysts than Ni for magnesium hydrides. Although Ni is commonly used for hydrogen absorption in Mg and its alloys, it is not a catalyst as good as Ti and V. This has been attributed to the effect of oxygen. Titanium and vanadium have very strong affinities to oxygen, and their oxides can not be reduced by hydrogen under normal conditions. This is the reason why Ni has better catalytic effects than Ti and V in conventional Mg-based alloys. Mechanical milling of MgH2 with Ti or V leads to titanium or vanadium hydrides, which could protect Ti or V from oxidation, and therefore the catalytic effect towards hydrogen is preserved. To understand the intrinsic mechanisms of 3d transition metal alloying effects on the dehydrogenating properties of magnesium hydride, YONG et al[5], calculated the total energy and the electronic structure of the solution of the 10% (mole fraction) alloying elements such as Fe, Ni, Cu, Ti into method. It is found that the alloying elements are considered to destabilize the magnesium hydride, and hence the dehydro- genation properties of MgH2 are improved. Based on the previous experimental works which the dehydrogenation properties of MgH2 can be improved by the addition of 3d transition metal , such as Ni, Co, Mn, Cu, Ti, Fe, V, but there was little effort to understand the intrinsic mechanisms of 3d transition metal for Sc, V, Cr, Mn, Co, Zn. Hence, in this paper, a first-principles plane-wave pseudopotential method based on the density functional theory has been used to investigate the energy and electronic structure of magnesium hydride (MgH2) alloyed by 3d transition metal elements. Moreover, the structural stability and electronic mechanism of 3d transition metal alloying magnesium hydride were analyzed and discussed, and then, some new results would be given.
2 Method of models of computation
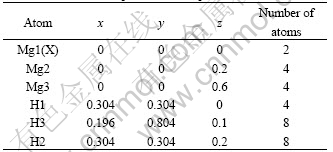
MgH2 has a tetragonal symmetry (P42/mnm, Group No.136). Two Mg atoms occupy (0, 0, 0) site and four H atoms locate in (0.304, 0.304,0) sites (Fig.1 (a)). The lattice parameters are a=0.450 1 nm and c=0.301 0 nm, respectively[6].In this work, we emplo- yed a supercell with 5 times in c axis of the unit cell, as shown in Fig.1(b). Due to the symmetry require- ment, there are three nonequivalent sites for magne- sium and hydrogen atoms in the supercells, as listed in Table 1. To investigate the influence of alloying elements X (X denotes 3d transition metal) on the structural stability, the total energy and electronic structure of magnesium hydrides, two Mg atoms in the supercells are replaced by alloying atoms. Hence, the Mg8X2H20 supercells are given.
Table 1 Structure of supercell used in present calculation
Cambridge serial total energy package (CASTEP) [7], a first-principles plane-wave psendopotentials method based on density functional theory, is used in this work. CASTEP uses a plane-wave basis set for the expansion of the single-particle Kohn-Sham wave- functions, and psendopotentials to describe the computationally expensive electron-iron interaction, in which the exchange-correlation energy by the generalized gradient approximation(GGA) of Perdew is adopted for all elements in our models by adopting Perdew- Burke-Ernzerhof parameters[8]. Ultrasoft represented in reciprocal space is used[9]. In the present calcu- lations, the cutoff energy of atomic wave functions (PWs), Ecut, is set at 310 eV. Sampling of the irredu- cible wedge of Brillouin zone is performed with a regular Monkhorst-Pack grid of special k-points, which is 6��6��6. A finite basis set correction and Pulay scheme of density mixing[10, 11] are applied for the evaluation of energy and stress. All atomic positions in our model have been relaxed according to the total energy and force using BFGS[12] scheme, based on the cell optimization criterion (RMs force of 0.5 eV/nm, stress of 0.1GPa, and displacement of 0.000 2 nm). The calculation of total energy and elec- tronic structure are followed by cell optimization with SCF tolerance of 2.0��10��6 eV.
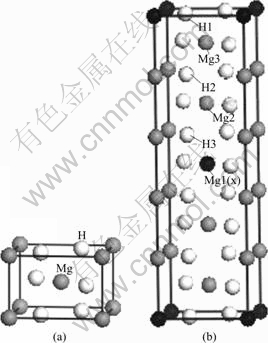
Fig.1 Models of atomic cells used in calculation: (a) Unit cell of MgH2; (b) Supercell of five times of unit cell of MgH2
3 Results and discussion
3.1 Equilibrium structure of crystal
The lattice constants of MgH2 are estimated from the minimized total energy, it can be found that the present lattice parameters a and c of MgH2 are 0.453 3 and 0.302 2 nm, respectively, which are close to the experimental values[6] of a=0.450 1 nm and c=
0.301 0 nm and are also in agreement with the results calculated by SONG et al[5]. The energy of its primitive cell is ��1 010.078 6 eV. The lattice para- meters of all the calculation Mg8X2H20 model are listed in Table 2. It can be found that with the addition of X , the equilibrium lattice constants (a) of all the supercell decrease, c also decreases, not including Sc and Ti. It can also be found that the unit cell volume of MgH2 reduces by alloying elements considered (not including Sc) in the present work.
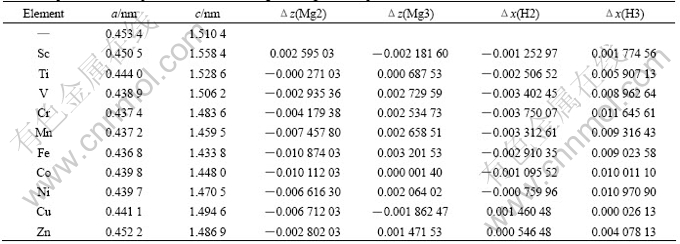
Analysis of the forces shows that the force on the alloying atom is F(X)=0, the forces on Mg2, Mg3, H1 atoms are zero in x and y directions, but nonzero in z direction, and the forces on H2��H3 atoms are zero in z direction, but nonzero in x and y directions .This indicates that internal relaxation of the atoms within the supercell is carried out for the considered systems. As required by group symmetry, the atoms X and H1 are fixed during the relaxation (Fig.1(b)). And the internal relaxation is performed in two steps. Firstly, Mg2 and Mg3 atoms are relaxed independently along z direction, while H2 and H3 atoms are kept at their initial positions. The variation of the atomic coordinates of Mg2 or Mg3 atoms is defined as ��z(Mg2/Mg3)=z��z0, where z0 is the initial atomic coordinate of Mg2 or Mg3 atom (Table 1). The final values of ��z(Mg2) and ��z(Mg3) are estimated as listed in Table 2. It can be found that the changes of the positions of Mg2 and Mg3 atoms in the z direction are very small due to the addition of alloying elements, owing to the fact that Mg2 and Mg3 atoms are the second nearest-neighbor atoms, resulting in a small energy difference between the relaxed and un-relaxed positions. Secondly, Mg2 and Mg3 atoms are fixed at their new positions, and H2 and H3 atoms are relaxed along x directions(As required by group symmetry, x=y) independently, with the change of ��x(H2/H3)= x��x0, where x0 is the initial atomic coordinate of H2 or H3 atoms in the supercell (Table 1). The final values of ��x(H2) and ��x(H3) are estimated and are listed in Table 2. It is found that the value of ��x(H2) is smaller than that of ��x(H3). This is consistent with the fact that H3 atom is the nearest neighbor of the alloying atom in the present supercell(Fig.1(b)). Therefore, from the structure relaxation study, it is noted that the alloying elements (not including Cu) mainly influence the position of the matrix atoms in their vicinity, i.e. only the positions of the nearest neighbor H3 atoms are significantly changed. The influence of alloying elements on the position of H3 atoms increases in an order of Sc, Zn, Ti, V, Fe, Mn, Co, Ni, Cr.
3.2 Heat of formation
Previous works show that the thermodynamic aspects of magnesium hydride formation/ decomposition are described by pressure-composition isotherms under the given temperature (T). The overall reaction of hydride formation is composed of three steps. Firstly, the host metal dissolves some hydrogen as a solid solution ( phase), keeping alloy structure unchanged. Secondly, when the hydrogen pressure altogether with concentration of H in the host metal is increased, the interactions between hydrogen atoms become locally strong, and the tetragonal structure hydride (
phase), keeping alloy structure unchanged. Secondly, when the hydrogen pressure altogether with concentration of H in the host metal is increased, the interactions between hydrogen atoms become locally strong, and the tetragonal structure hydride ( phase) becomes nucleation and growth. While two phases coexist, the isotherms appear a flat plateau, the length of which means that how much H2 can be stored with small pressure variations, and that the plateau or equilibrium pressure depends strongly on temperature and is related to the changes of enthalpy (
phase) becomes nucleation and growth. While two phases coexist, the isotherms appear a flat plateau, the length of which means that how much H2 can be stored with small pressure variations, and that the plateau or equilibrium pressure depends strongly on temperature and is related to the changes of enthalpy (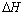 ) and of (
) and of (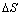 ) entropy, respectively. Finally, the concentration of H in the host metal show no further change if hydrogen pressure is kept on increasing. The reaction of hydride decomposition is opposite to hydride formation, but exists in time retardation compared with hydride formation. During the second step, it is often related to the equilibrium
) entropy, respectively. Finally, the concentration of H in the host metal show no further change if hydrogen pressure is kept on increasing. The reaction of hydride decomposition is opposite to hydride formation, but exists in time retardation compared with hydride formation. During the second step, it is often related to the equilibrium
Table 2 Equilibrium lattice parameters and total energies of Mg8X2H20 supercell model
hydrogen pressure through Van��t Hoff equation[13]:
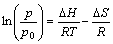 (1)
(1)
where P is flat plateau pressure for decomposition, P0 is the standard pressure (=0.1 MPa), is the heat of formation, is the entropy change, T is the temperature and R is the gas constant. The entropy change in Eqn.(1) is dominated by the entropy loss of the gaseous hydrogen, roughly 130.8 J/mol[13] for MgH2 under consideration. Hence, Eqn.(1) can be expressed as
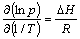 (2)
(2)
From Eqn.(2) it can be found that the heat of formation () of magnesium hydride determines the flat plateau pressure of magnesium hydride decomposition under the given temperature(T).The smaller the heat of formation () is, the lower the pressure is, which indicates the enhanced dehydrogenating properties of magnesium hydride[5, 14]. Hence, one may only concentrate on the heat of formation in order to understand the dehydrogenating properties of magnesium hydride.
Based on the above summary, the heat of formation of MgH2 can be calculated by[5]
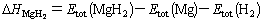 (3)
(3)
where Etot(MgH2) is the energy of primitive cell of MgH2 , Etot(Mg) is the single atomic energy of HCP- Mg in the solid states, and Etot(H2) is the total energy of hydrogen molecule. The calculation energies of Mg atoms are also listed in Table 1 using the same code as primitive cell model. The total energy of hydrogen molecule is calculated as ��2.320Ry (�֨C31.565 2 eV)[13], using von Barth-Hedin exchange- correlation potential. The present heat of formation of MgH2 is �C62.30 kJ/mol, slightly bigger than the value �C73.5 kJ/mol from the thermodynamic data under the given temperature (T��673 K) measured experimentally by Bogdanov��? et al[15].
In order to investigate the influence of 3d transition metal alloying elements on the structural stability of magnesium hydride, a parameter 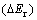 is used to estimate the relative stability of alloyed magnesium hydrides compared with that of MgH2. Then,
is used to estimate the relative stability of alloyed magnesium hydrides compared with that of MgH2. Then, 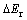 is calculated by[5]
is calculated by[5]
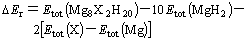 (4)
(4)
where the first two terms on the right-hand side of Eqn.(4) are the total energy of MgH2��X with the current supercell and primitive cell of MgH2, respectively, 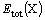 denotes the total energy of element X and is calculated using the same code as for MgH2��X. In our calculations, FCC-type structure is used for Mn, Ni, Cu and Zn, respectively, BCC-type structure is used for V, Cr and Fe��respectively, HCP-type structure is used for Sc, Ti, Co and Mg, respectively. The values of
denotes the total energy of element X and is calculated using the same code as for MgH2��X. In our calculations, FCC-type structure is used for Mn, Ni, Cu and Zn, respectively, BCC-type structure is used for V, Cr and Fe��respectively, HCP-type structure is used for Sc, Ti, Co and Mg, respectively. The values of 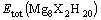 and calculated here are listed in Table 3. It is found that value in our calculations for MgH2��Ti system is close to that given by SONG et al[5], but the other values are smaller than those given by SONG et al[5] for MgH2��Fe, MgH2��Ni, MgH2��Cu, respectively. The data in Table 3 also show that the negative heat of formation of magnesium hydride alloyed by X (not including Sc) decreases in comparison with that of MgH2, and the amount of reduction increases in an order of Fe, Ti, Co, Zn, Ni, V, Mn, Cu and Cr. It is noted that the values for MgH2��Cu and MgH2��Cr are very close to the absolute value of the heat of formation of MgH2, implying the two alloyed systems are more destabilized. Moreover, value for MgH2��Ni is bigger than that of MgH2��Ti, which is against the experiment result[4]. In fact, some alloying elements(such as Ti, V, Fe, Co, Ni and Cu) considered here can form compounds with Mg and hydrogen, for example, TiH2, VH0.81, MgCu2[1, 2], or form complex magnesium hydrides, such as Mg2FeH6, Mg2CoH5, Mg2NiH4 and MgCu2[3, 4]. The influence of alloying elements considered here on the structural stability of MgH2 should be addressed by the formation of these second phases, which can be found elsewhere.
and calculated here are listed in Table 3. It is found that value in our calculations for MgH2��Ti system is close to that given by SONG et al[5], but the other values are smaller than those given by SONG et al[5] for MgH2��Fe, MgH2��Ni, MgH2��Cu, respectively. The data in Table 3 also show that the negative heat of formation of magnesium hydride alloyed by X (not including Sc) decreases in comparison with that of MgH2, and the amount of reduction increases in an order of Fe, Ti, Co, Zn, Ni, V, Mn, Cu and Cr. It is noted that the values for MgH2��Cu and MgH2��Cr are very close to the absolute value of the heat of formation of MgH2, implying the two alloyed systems are more destabilized. Moreover, value for MgH2��Ni is bigger than that of MgH2��Ti, which is against the experiment result[4]. In fact, some alloying elements(such as Ti, V, Fe, Co, Ni and Cu) considered here can form compounds with Mg and hydrogen, for example, TiH2, VH0.81, MgCu2[1, 2], or form complex magnesium hydrides, such as Mg2FeH6, Mg2CoH5, Mg2NiH4 and MgCu2[3, 4]. The influence of alloying elements considered here on the structural stability of MgH2 should be addressed by the formation of these second phases, which can be found elsewhere.
3.3 Electronic structure
3.3.1 Density of states
Total and partial density of states (DOS) for MgH2 are performed as shown in Fig.2. The total DOS shows a few main bonding peaks in the energy range between Fermi level and �C7.0 eV. It is found that the main bonding peaks (between Fermi level and �C3.0 eV) mainly originates from the contribution of valence electron numbers of H(s), Mg(p) and little Mg(s) orbitals, while the main bonding peaks between �C4.0 and �C3.0 eV are the result of the bonding among H(s), Mg(s) and little Mg(p). The main bonding peaks between �C7.0 and �C4.0 eV are caused by H(s) and Mg(s).The total DOS in Fig.2 shows that the highest bonding possesses the characteristic of significant s-p hybridization, contributed by the hydrogen s and magnesium p and s electrons. This leads to a relatively high formation energy of MgH2.This aspect was changed in the alloyed 3d transition metal MgH2 systems.
The alloying elements considered here can be classified into three groups: 1) Sc, Ti, V, Cr and Mn; 2) Fe, Co and Ni; 3) Cu and Zn. According to experimental findings[1-4], in group 1, Ti and V do not form compounds with Mg, but can form hydrides TiH2 and VH0.81[1, 2], respectively. For group 2, Fe, Co and Ni can form hydrides Mg2FeH6, Mg2CoH5 and Mg2NiH4[3, 4], respectively. For group 3, Cu can form hydrides MgCu2[2] with Mg. These features are determined by the electronic structure.
Table 3 Energy of pure metal atom, total energies of Mg8X2H20 supercell model and parameter
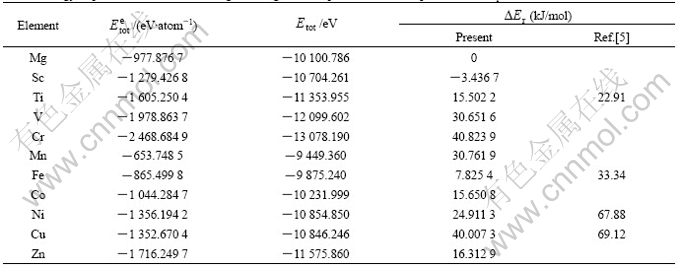
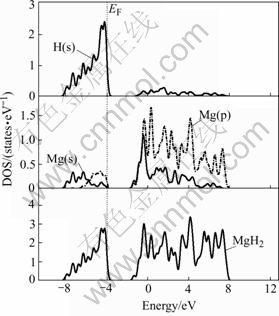
Fig.2 Total and partial density of states of MgH2 crystal
1) Sc, Ti, V, Cr, Mn
Fig.3(a) shows the total and partial density of states (DOS) of MgH2 supercell with alloying system such as Sc, Ti, V, Cr, Mn. In MgH2-Ti (Fig.3(c)), the bonding peaks near Fermi energy are contributed by
Ti(d), the main part, and a weak Mg(s), Mg(p) and H(s) electrons. The s-p bonding peaks between magnesium and hydrogen appear in the energy range between �C2.0 and �C6.0 eV, there is also weak contribution of Ti(d) electrons to the bonding peaks. The s electron between magnesium and hydrogen appears in the energy region from �C6.0 to �C9.0 eV. Total and partial density of states (DOS) of MgH2��X(X= Sc, V, Cr, Mn) systems are similar to those of MgH2��Ti. In MgH2��Sc(Fig.3(b)), there is a contribution of the Mg(s), Mg(p), H(s) and a little Sc(d) electrons to the bonding peaks from �C2.5 to �C7.0 eV, Mg(s) and H(s) electrons bonding appears in the energy region from �C7.0 to �C9.0 eV. In MgH2��V (Fig.3(d)), the bonding peaks between �C2.0 eV and �C6.0 eV is contributed by Mg(s), Mg(p), H(s) and a little V(d) electrons, while these peaks between �C6.0 eV and �C9.0 eV is the result of the interaction between Mg(s) and H(s) electrons.For MgH2��Cr (Fig.3(e)) and MgH2��Mn(Fig.3(f)), the bonding peaks (between �C1.5 eV and �C6.0 eV) are caused by the bonding of Mg(s), Mg(p), H(s) and a little Cr(d) or Mn(d) electrons. The s electron bonding between magnesium and hydrogen all appears in the energy region from �C6.0 to �C9.0 eV for MgH2��X (Sc, Ti, V, Cr, Mn) systems.
2) Fe, Co, Ni
Figs.4(a), (b) and (c) show the total and partial density of states (DOS) of MgH2��Fe, MgH2��Co, MgH2��Ni, respectively. In Fig.4(a), it can be found that the bonding peaks of total DOS in the energy range from �C0.5 to �C1.5 eV show Fe(d) electrons characteristics, while a little Mg(s), Mg(p), H(s) electrons also contribute to some extent. The highest bonding peak in total DOS is located at about �C1.0eV below Fermi energy. These feature implies that there is a relatively weak bonding between Fe, Mg and H in the MgH2��Fe systems. The other bonding peaks in total DOS are located in the energy range from �C2.0 to �C9.0 eV below Fermi energy. These peaks are the results of the interactions between Mg and H. As far as the highest bonding peak in total DOS at about �C4.0 eV below the Fermi energy is concerned, it is caused by the bonding of Mg, H1 and H3. In MgH2��Co( Fig.4(b)), the bonding peaks near Fermi energy are dominated by the strong Co(d), and a weak Mg(s), Mg(p) and H(s) electrons, the highest bonding peak in total DOS is located at about �C2.0 eV below Fermi energy, while the second bonding peak at about �C5.0 eV below Fermi energy is the results of the interactions of Co, H1 and H3. In MgH2��Ni ( Fig.4(c)), there is a contribution of the main Ni(d) and a weak Mg(s), Mg(p) and H(s) electrons to the bonding peaks near Fermi energy. The highest bonding peak in total DOS is located at about �C2.5 eV below Fermi energy. While the bonding peak at about �C5.0 eV below the Fermi energy is the results of the interactions between H1(s), H3(s), Mg2(s), Mg3(s), Mg2(p) and Mg3(p), this indicates a relatively stronger interaction between Mg3 and H1 atoms. In partial DOS, the magnitude of the Mg(p) electron peak in the energy range from �C2.0 to �C4.0 eV is reduced compared with that of MgH2, which means a relatively weak interaction between Mg and H atoms.
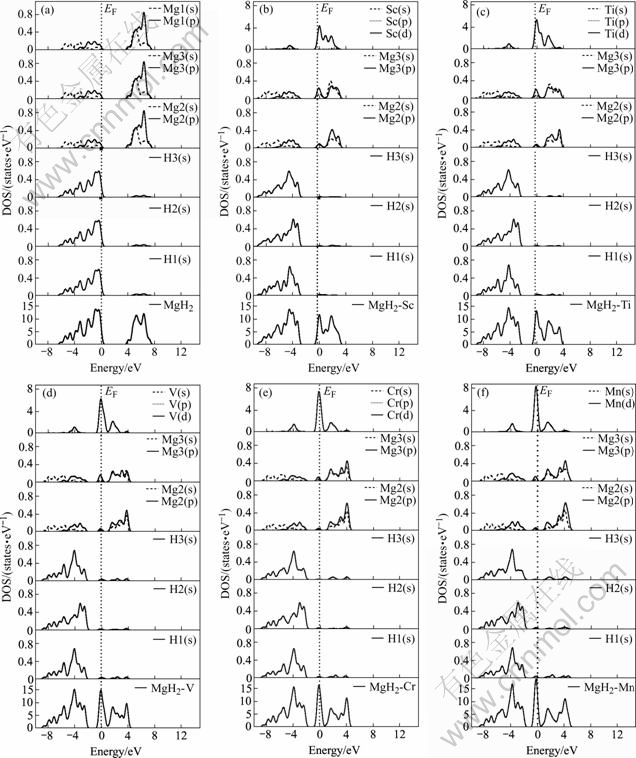
Fig.3 Total and partial density of states of MgH2(a), MgH2��Sc(b), MgH2��Ti(c), MgH2��V(d), MgH2��Cr(e) and MgH2��Mn(f)
3) Cu, Zn
Fig.5 shows the total and partial density of states (DOS) of MgH2��Cu and MgH2��Zn. In Fig.5(a), the main bonding peaks near Fermi energy are contributed by Cu(d), Mg(p) and H(s) electrons. The contribution of H2(s) electron to the peak is smaller than that of the other two H atoms. The highest bonding peak, located at �C3.0 eV, shows the characteristics of Cu(d) electrons, but it also contains the contributions of Mg2(p) and H2(s) electrons. The other peaks are caused by the bonding of Mg2(p), Mg3(p), H1(s), H3(s), with some level of the Cu(d) electrons. In MgH2��Zn (Fig.5(b)), the main bonding peaks near Fermi energy are dominated by Zn(d), Mg(p) and H(s) electrons, while the highest bonding peak, located at �C6.0 eV, is the result of the bonding between main Zn(d) and weak Mg(s) and H(s) electrons. The other peaks are dominated by Mg2(p), Mg3(p), H1(s) and H3(s) electrons.
3.3.2 Bonding and charge density distribution
In order to understand the intrinsic mechanism of the bonding and influence of 3d transition metal alloying on the structural stability of MgH2, a charge density analysis was carried out on (1 1 0) plane. Fig.6 shows the charge distribution on (1 1 0) plane of MgH2 and the considered alloying systems. Fig.6(c) shows the charge distribution of MgH2��Ti systems. Ti��H1 and Ti��H3 bonds can be identified, while Mg3��H2, and Mg2��H3 , Mg2��H2, and Mg3��H3 bonds are similar to the case of MgH2, but Mg2��H2, and Mg3��H3 bonds slightly reduces. The bonds of MgH2��Sc, MgH2��V, MgH2��Cr, MgH2��Mn systems are similar to that of MgH2-Ti, which can be seen in Figs.6(b), 6(d), 6(e), 6(f), respectively. For MgH2��Fe(Fig.6(g)), MgH2��Co(Fig.6(h)) and MgH2��Ni (Fig.6(i)), the bond strength between Mg and H atoms is reduced by addition of Fe, Co, or Ni. The charge distribution on (110) plane of MgH2��Cu, MgH2��Zn systems is shown as Fig.6(j), 6(k), respectively. It is found that the distributions of electrons between Mg and H atoms are similar to that in the MgH2 systems by addition of Cu or Zn. Only the Mg2��H2 bond are slightly weakened.
3.4 Dehydriding properties
The influence of alloying elements on the dehydrogenation properties of MgH2 was investigated experimentally, such as Ni, Co, Mn, Cu, Ti, Fe and V. It is found that the dehydrogenation properties are improved by the addition of above these alloying elements. For MgH2-Ti system, due to the formation of the second phase TiH2[2], the hydrogen atoms is held by Ti atoms. However, DOS (Fig.3(c)) and the charge distribution on (110) plane (Fig.6(c)) of the system show that the bonding strength between Mg and H atoms is slightly reduced, compared with that of MgH2, and the unit cell volume is also reduced in this system(Table 2).Hence, the structural stability of MgH2 is reduced, then the dehydrogenation properties of MgH2 is improved by addition of titanium. The alloying element V, Mn, shows a similar effect to Ti due to their similar electronic structure features shown Figs.4(d),.4(e) and 8(d), 8(e), respectively. But for MgH2��Cr systems, the positions of the nearest neighbor H3 atoms with alloy atoms are significantly changed(Table.2), which indicates the strongest bond between Cr and H3 exist and this lead to the weaker bond between Mg and H. Hence, the dehydrogenation properties of MgH2 is significantly improved by addition of Cr. For MgH2��Sc systems, the dehydrogenation properties of MgH2 is not be improved, it could be caused by unit cell volume increased[13], which should be further studied. For MgH2��Fe and MgH2��Co systems, the stability of the considered system is reduced by 7.825 4 kJ/mol and 15.650 8 kJ/mol(Table 3), respectively. Weak bonding peak near the Fermi energy were found in DOS (Figs.4(a) and 4(b)) and the charge distributions on (110) plane(Figs.6(g) and 6(h)) also show a reduction of the electron density between Mg and H atoms. Therefore, the dehydrogenation kinetics of MgH2 is improved by addition of Fe or Co. For MgH2��Ni systems, the dehydrogenation kinetics of MgH2 is significantly improved compared with that of MgH2��Fe and MgH2��Co by addition of Ni as mentioned above(see Table 3). The structural stability of the considered system is reduced by 24.911 3 kJ/mol, which can be expected due to the fact that means that the formation of the second phase Mg2NiH4 with a heat of formation of ��62.7 kJ/mol H2[16], larger than that of MgH2. This indicates that the H atoms in the mixture of MgH2 and Mg2NiH4 compounds can be released more readily than those in MgH2. Hence, The structural stability of MgH2��Ni systems is lower than that of MgH2��Fe and MgH2��Co systems. For MgH2��Cu systems, the parameter () is close to the absolute value of the heat formation of MgH2. It is found that the weaker bonds between Mg and H2 is seen as Figs.5(a) and 6(j). And due to the formation of MgCu2, Mg atoms are completely held by Cu atoms. Hence, the previously engaged H atoms can be readily released from MgH2��Cu systems, leading to the improvement of the dehydrogenation properties of MgH2. the alloying elements Zn shows a similar effect to Cu by the analysis the electronic structure shown in Figs.5(b) and 6(k), but Mg atoms are not completely held by Zn atoms, which can be seen in Fig.5(b).
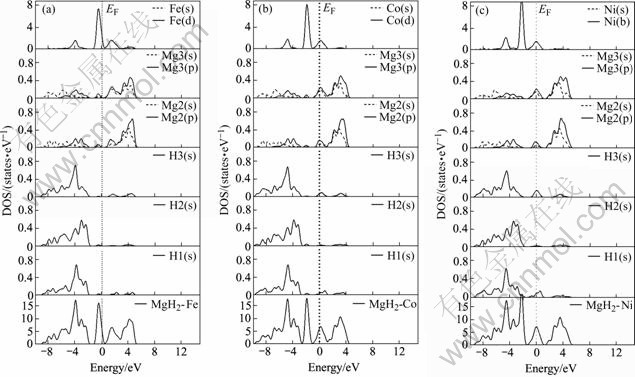
Fig.4 Total and partial density of states of MgH2��Fe(a), MgH2��Co(b) and MgH2��Ni(c)
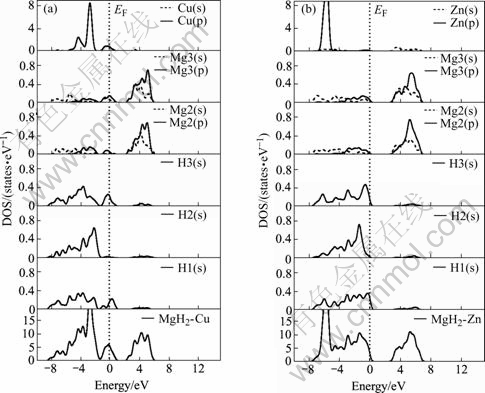
Fig.5 Total and partial density of states of MgH2��Cu(a) and MgH2��Zn(b)
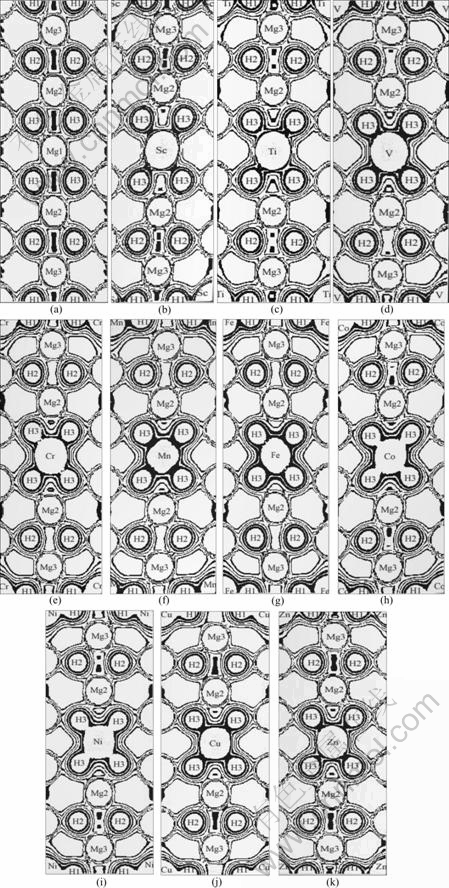
Fig.6 Charge distribution on (110) plane of MgH2 (a), MgH2��Sc(b), MgH2��Ti(c), MgH2��V(d), MgH2��Cr(e), MgH2��Mn(f), MgH2��Fe(g), MgH2��Co(h), MgH2��Ni(i), MgH2��Cu(j) and MgH2��Zn(k)
4 Conclusions
1) From the relaxation study, it is noted that the alloying elements (not including Cu) mainly influence the position of the matrix atoms in their vicinity: i.e., only the positions of the nearest neighbor H3 atoms are significantly changed. The influence of alloying elements on the position of H3 atoms increases in an order of Sc, Zn, Ti, V, Fe, Mn, Co, Ni and Cr.
2) Through calculations of the negative heat formation of magnesium hydride alloyed by X (X denotes 3d transition metal) element, it is found that when a little X (not including Sc) dissolve into magnesium hydride, the structural stability of alloying systems decreases, which indicates that the dehydrogenation properties for MgH2 is improved.
3) After comparing the densities of states (DOS) and the charge distribution of MgH2 with and without X alloying, it is found that the improvement for the dehydrogenation properties of MgH2 alloyed by X attributes to the fact that the weakened bonding between magnesium and hydrogen is caused by the stronger interactions between X (not including Cu) and hydrogen.
4) For MgH2��Cu systems, the parameter  is close to the absolute value of the heat formation of MgH2. This may mainly be due to the formation of MgCu2, in which Mg atoms are completely held by Cu atoms, and the previously engaged H atoms can be readily released from MgH2��Cu systems. But for MgH2��Cr systems, the positions of the nearest neighbor H3 atoms with alloy atoms are significantly changed, which means that the strongest bond between Cr and H3 exist and this lead to the weaker bond between Mg and H. Therefore, the dehydrogenation properties for MgH2 is significantly improved by addition of Cu or Cr.
is close to the absolute value of the heat formation of MgH2. This may mainly be due to the formation of MgCu2, in which Mg atoms are completely held by Cu atoms, and the previously engaged H atoms can be readily released from MgH2��Cu systems. But for MgH2��Cr systems, the positions of the nearest neighbor H3 atoms with alloy atoms are significantly changed, which means that the strongest bond between Cr and H3 exist and this lead to the weaker bond between Mg and H. Therefore, the dehydrogenation properties for MgH2 is significantly improved by addition of Cu or Cr.
5) The calculation results of the improvement for the dehydrogenation properties for MgH2��X (X=Ti, V, Mn, Fe, Co, Ni, Cu) systems are in agreement with the experimental results. The dehydrogenation properties for MgH2 are expected to be improved by addition of Cr, Zn alloying elements.
Acknowledgment
We wish to thank Dr.Y.Song, Professor of Department of Materials, Queen Mary, University of London, for his helpful discussion.
References
[1] Liang G, Huot J, Boily S, et al. Hydrogen desorption kinetics of a mechanically milled MgH2+5at.%V nanocomposite[J]. J Alloys Compd, 2000, 305: 239��245.
[2] Shang C X, Bououdina M, Song Y, et al. Mechanical alloying and electronic simulations of (MgH2+M) systems (M=Al,Ti,Fe,Ni,Cu and Nb) for hydrogen storage[J]. In J Hydrogenenergy, 2004, 29: 73��80.
[3] Huot J, Hayakawa H, Akiba E. Preparation of the hydrides Mg2FeH6 and Mg2CoH5 by mechanical alloying followed by sintering[J]. J Alloys Compd, 1997, 248: 164��167.
[4] Liang G, Huot J, Boily S, et al. Catalytic effect of transition metals on hydrogen sorption in nanocrystalline ball milled MgH2-Tm (Tm=Ti, V, Mn, Fe and Ni) systems[J]. J Alloys Compd, 1999, 292: 247��252.
[5] Song Y, Guo Z X, Yang R. Influence of selected alloying elements on the stability of magnesium dihydride for hydrogen storage applications: A first-principles investigation[J]. Phys Rev B, 2004, 69: 094205��094215.
[6] Bortz M, Bertheville B, Bottqer G, et al. Structure of the high pressure phase
Structure of the high pressure phase 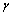 -MgH2 by neutron powder diffraction[J]. J Alloys Compd, 1999, 287: L4��6.
-MgH2 by neutron powder diffraction[J]. J Alloys Compd, 1999, 287: L4��6.
[7] Segall M D, Lindan P L D, Probert M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys:Condens Matter, 2002, 14: 2717��2744.
[8] Marlo M, Milman V. Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals[J]. Phys Rev B, 2000, 62: 2899��2907.
[9] Vanderbilt D. Soft self-consistent pseudopotentitals in a generalized eigenvalue formalism[J]. Phys Rev B, 1990, 41: 7892��7895.
[10] Hammer B, Hansen L B, Norkov J K. Improved adsorption energetics withen density-functional theory using revised Perdew-Burke-Ernzerhof functionals[J]. Phys Rev B, 1999, 59: 7413��7421.
[11] Franscis G P, Payne M C. Finite basis set corrections to total energy pseudopotential calcaulations[J]. J Phys: Condens Matter, 1990, 2: 4395��4404.
[12] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations[J]. Phys Rev B, 1976, 13: 5188��5192.
[13] Nakamura H, Nguyen-Manh D, Pettifor D G. Electronic structure and energetics of LaNi5, ��-La2Ni10H and ��-La2Ni10H14[J]. J Alloys Compd, 1998, 281: 81��91.
[14] Nambu T, Ezaki H , Yukawa H, et al. Electronic structure and hydriding property of titanium compounds with CsCl-type structure[J]. J Alloys Compd, 1999, 293��295: 213��216.
[15] Bogdanov��? B, Bohmhammel K, Christ B, et al. Thermodynamic investigation of the magnesium-hydrogen system[J]. J Alloys Compd, 1999, 282: 84��92.
[16] Yamaguchi M, Akiba E. Electronic and magnetic properties of metal and ceramics part(��)[A]. Cahn r w, Haasen p, Kramer e j. Materials Science and Technology[C]. Weinhein: VCH, 1994. 333.
Foundation item: Project(20020530012) supported by Doctoral Program Foundation of Ministry of China.
Corresponding author: ZHOU Dian-wu; Tel: +86-13017297124; E-mail: ZDWe_mail@163.net
(Edited by LONG Huai-zhong)

