Œƒ’¬±‡∫≈£∫1004-0609(2010)05-0969-07
◊ºæß”Î∆‰Ω¸À∆œ‡ƒ£ƒ‚÷–µƒ ∆∫Ø ˝
ø◊œÈ¥∫£¨∫˙Õ˚”Ó£¨µÀª‘«Ú
(∫˛ƒœ¥Û—ß ŒÔ¿Ì”ÎŒ¢µÁ◊”ø∆—ß—ß‘∫£¨≥§…≥ 410082)
’™ “™£∫‘≠◊”º‰œ‡ª•◊˜”√ ∆∫Ø ˝ «¥”‘≠◊”≥fl∂»…œ∂‘≤ƒ¡œ∏˜÷÷Ãÿ–‘Ω¯––º∆À„ª˙ƒ£ƒ‚—–æøµƒª˘¥°°£”…”⁄◊ºæߺ∞∆‰Ω¸À∆œ‡Ãÿ ‚µƒº∏∫Œππ–Õ£¨∆‰≤…”√µƒœ‡ª•◊˜”√ ∆µƒ÷˜“™Ãÿµ„ «∞¸∫¨¥´Õ≥ ∆∫Ø ˝÷–√ª”–µƒ’Òµ¥œÓ°£◊ºæßÃÂœµ ∆∫Ø ˝¥ÛÃÂ∑÷Œ™¡Ω¿‡£∫“ª¿‡ «“‘Dzugutov ∆∫ÕLJG ∆Œ™¥˙±Ìµƒª˘”⁄¥´Õ≥ ∆∫Ø ˝µƒ◊˜”√ ∆°£’‚¡Ω÷÷ ∆≥£”√”⁄µ•‘≠◊”ÃÂœµ”ÎÀ´‘≠◊”ÃÂœµµƒ◊ºæßÃÂœµƒ£ƒ‚£¨Dzugutov ∆ «“ª÷÷∂Ã≥Ãœ‡ª•◊˜”√£¨À¸«„œÚ”⁄–Œ≥…æ€Àƒ√Êã¨≥£”√”⁄≤£¡ß赃ÃÂœµ÷–°£LJG ∆º”«ø¡À≥§≥Ãœ‡ª•◊˜”√£¨ «ºÚµ•◊ºæßÃÂœµƒ£ƒ‚÷–”¶”√◊Óπ„∑∫µƒœ‡ª•◊˜”√ ∆°£¡Ì“ª¿‡ «“‘Moriarty-Widom ∆”ÎForce Match∑Ω∑® ∆Œ™¥˙±ÌµƒRealistic–Õ ∆∫Ø ˝£¨’‚÷÷ ∆∫Ø ˝ª˘”⁄µ⁄“ª–‘‘≠¿Ìº∆À„ªÚ µ—ÈΩ·π˚£¨‘⁄∂‡‘™∫œΩ◊ºæßÃÂœµƒ£ƒ‚÷–”–π„∑∫µƒ”¶”√°£÷˜“™ΩÈ…‹“‘…œ¡Ω¿‡ ∆∫Ø ˝£¨≤¢∂‘∆‰Ãÿµ„Ω¯––∆¿ ˆ°£
πÿº¸¥ £∫‘≠◊”º‰œ‡ª•◊˜”√ ∆£ª◊ºæߣª ∆∫Ø ˝£ª◊ºæß ∆∫Ø ˝£ªRealistic–Õ ∆∫Ø ˝£ªº∆À„ª˙ƒ£ƒ‚
÷–Õº∑÷¿‡∫≈£∫O 469°°°° Œƒœ◊±Í÷æ¬Î£∫A
Interatomic potentials for simulation of
quasicrystal and its approximant
KONG Xiang-chun, HU Wang-yu, DENG Hui-qiu
(College of Physics and Micro-electronic Science, Hunan University, Changsha 410082, China)
Abstract: Interaction potentials are essential in the atomic-scale computer simulation for materials properties. In contrast to the regular potentials, the potential functions for quasicrystal and its approximant have many local minimums due to its particular topology. There are mainly two kinds of potentials for quasicrystal. One is the potentials based on regular ones represented by Dzugutov potential and LJG potential. They are generally applied in the simulation of monatomic and diatomic systems. Dzugutov potential is a short-term interaction which strongly favors polytetrahedral clusters, and it is frequently applied in the simulation of glasses. LJG potential strengthens the long-range interaction compared to Dzugutov potential. It is the most widely used interaction in the simulation of simple systems. The other kind is Realistic potentials represented by Moriarty-Widom potential and potentials from Force Match method. These potentials are based on the first principle and experimental results, which plays an important role in the simulation of alloy systems such as AlNiCo. Those potentials are reviewed and some remarks are given.
Key words: interatomic potential; quasicrystal; potential function; quasicrystal potential function; Realistic potential function; computer simulation
ÀÊ◊≈º∆À„ª˙»Ì”≤º˛µƒøÏÀŸ∑¢’π£¨º∆À„ª˙ƒ£ƒ‚‘⁄œ÷¥˙≤ƒ¡œ—ß°¢ŒÔ¿Ì—ß∫ժؗߵ»—ßø∆—–æøµ±÷–∆µΩ‘Ω¿¥‘Ω÷ÿ“™µƒ◊˜”√£¨÷Ω•≥…Œ™”ο̬€—–æø∫Õ µ—È—–æøº∏∫ıÕ¨µ»÷ÿ“™µƒ—–æø ÷∂Œ°£º∆À„ª˙ƒ£ƒ‚“ª∑Ω√ʃ‹¥”Œ¢π€≥fl∂»…œ∫‹∫√µÿΩ‚ Õƒ≥–©“—”– µ—ÈΩ·π˚£¨¡Ì“ª∑Ω√Ê”÷ƒ‹∏˘æ›“—”–÷™ ∂¿¥‘§≤‚ƒ≥–©≤ƒ¡œµƒŒÔ¿Ì°¢ªØ—ß∫Õ¡¶—ß–‘ƒ‹µ»°£º∆À„ª˙ƒ£ƒ‚÷–◊Ó≥£”√µƒ «∑÷◊”∂Ø¡¶—ß∑Ω∑®∫Õ√…Ãÿø®¬Â∑Ω∑®°£‘⁄ª˘”⁄‘≠◊”≥fl∂»…œµƒº∆À„ª˙ƒ£ƒ‚π˝≥Ã÷–£¨◊Óπÿº¸µƒ «—°‘Ò∫œ µƒ‘≠◊”º‰œ‡ª•◊˜”√ ∆∫Ø ˝°£Õ®≥£ π”√µƒ‘≠◊”º‰œ‡ª•◊˜”√ ∆÷˜“™”–¡Ωà∆(¿˝»ÁLennard- Jones ∆[1])°¢»˝Ã ∆(¿˝»ÁTersoff ∆[2])∫Õ∂‡Ã ∆(¿˝»ÁEmbedded Atom Method£¨EAM ∆[3-4])°£¡Ωà∆“ª∞„”…Œ¸“˝œÓ∫Õ≈≈≥‚œÓ¡Ω≤ø∑÷◊È≥…£¨À¸Ωˆøº¬«¡Ω∏ˆ‘≠◊”º‰µƒœ‡ª•◊˜”√£¨∂¯∫ˆ¬‘¡À‘≠◊”º‰µƒ∂‡ÃÂ◊˜”√–ß”¶°£’‚¿‡ ∆±»Ωœ ”√”⁄√‹∂—Ω·ππ∫Õ‘≠◊”º‰ªÚÕ≈¥ÿº‰µÁ∫…÷ÿµ˛Ωœ…Ÿµƒ«È–Œ[5]°£»˝Ã ∆÷–Õ®π˝º”»Î”κ¸º–Ω«”–πÿµƒœÓ¿¥øº¬«»˝ÃÂœ‡ª•◊˜”√£¨≥…π¶µÿ√Ë ˆ¡ÀC°¢Si°¢Geµ»‘™ÀÿªÚ∆‰◊È≥…µƒπ≤º€ªØ∫œŒÔµƒÃÿ–‘°£“‘EAM ∆Œ™¥˙±Ìµƒ∂‡Ã ∆£¨ƒ‹Ωœ∫√µÿ√Ë ˆæ¯¥Û≤ø∑÷Ω Ù‘™Àÿº∞∆‰∫œΩµƒ∏˜÷÷–‘÷ [6]°£∂‘”⁄º∆À„ª˙ƒ£ƒ‚÷–’‚–©≥£”√ ∆∫Ø ˝µƒÃÿµ„º∞∆‰‘⁄æßÃÂ∫Õ∑«æß≤ƒ¡œ÷–µƒ”¶”√«Èøˆ£¨Œƒœ◊[5, 7-10]÷–“—”–≤ª…Ÿ∫‹∫√µƒ◊€ ˆ°£µ´ «‘⁄æßÃÂ∫Õ∑«æß÷ÆÕ‚µƒ◊ºæߺ∞∆‰Ω¸À∆œ‡≤ƒ¡œ£¨”…”⁄∆‰Ãÿ ‚µƒ‘≠◊”º∏∫Œ≈≈¡–Ãÿ’˜∫ÕµÁ◊”–‘÷ £¨Õ®≥£”¶”√µƒ ∆∫Ø ˝‘⁄¥¶¿Ì◊ºæß≤ƒ¡œ ±∂ºª·”ˆµΩ“ª∂®µƒ¿ßƒ—°£
”√”⁄◊ºæßÃÂœµµƒ ∆∫Ø ˝”Î√Ë ˆ¥´Õ≥æßè∫Õ∑«æßèÃÂœµµƒ ∆∫Ø ˝æfl”–≤ªÕ¨µƒÃÿµ„£¨Ω¸º∏ƒÍ¿¥µ√µΩ∫‹¥Ûµƒ∑¢’𰣑⁄¥À£¨±æŒƒ◊˜’fl∑÷¿‡¡–æŸΩ¸ƒÍ¿¥‘⁄◊ºæߺ∞∆‰Ω¸À∆œ‡ƒ£ƒ‚π˝≥Ã÷–≥£”√µΩµƒº∏÷÷‘≠◊”º‰œ‡ª•◊˜”√ ∆∫Ø ˝£¨≤¢∂‘∆‰∫Ø ˝Ãÿµ„∫Õµ‰–Õ”¶”√«ÈøˆΩ¯––∆¿ ˆ°£
1 ◊ºæ߃£ƒ‚ ∆∫Ø ˝µƒ“™«Û∫ÕÃÿµ„
¥”Ω·ππ…œø¥£¨¥´Õ≥æßÃÂ∞¸∫¨∆Ω“∆∂‘≥∆–‘∫Õ–˝◊™∂‘≥∆–‘£¨ÃÂœµ‘≠◊”≈‰Œªª∑æ≥œ‡∂‘ºÚµ•£¨¥”ƒ‹¡ø…œ±Ìœ÷Œ™Œ»∂®µƒµÕƒ‹Ã¨°£∂¯∏¥‘”µƒ◊ºæß∫œΩµ±÷–÷ª∞¸∫¨–˝◊™∂‘≥∆–‘∂¯√ª”–∆Ω“∆∂‘≥∆–‘£¨Ω·ππ÷–∞¸∫¨¥´Õ≥æßõ±÷–≤ª¥Ê‘⁄µƒŒÂ÷ÿ∂‘≥∆Ω·ππ£¨“Ú¥À£¨–Ë“™ΩË÷˙∂‡”⁄“ª÷÷µƒª˘±æΩ·ππ¿¥∆ì˙’˚∏ˆø’º‰£¨±Ìœ÷Œ™ƒ≥∏ˆªÚ∂‡∏ˆ∑ΩœÚ…œ≥ˆœ÷◊º÷‹∆⁄–‘≈≈¡–“‘º∞∏ª∫¨ŒÂ÷ÿ∂‘≥∆µƒ∂˛ Æ√ÊÃÂÕ≈¥ÿ◊”Ω·ππ£¨Œ™ƒ‹¡øΩœ∏flµƒ—«Œ»Ω·ππ°£”…”⁄◊ºæߔΥ´Õ≥æßÑ⁄Ω·ππ∫Õƒ‹¡øµ»∑Ω√ʥʑ⁄Ωœ¥Ûµƒ≤Ó“Ï£¨“Ú¥À£¨‘⁄◊ºæ߃£ƒ‚π˝≥Ã÷–≤…”√µƒ ∆∫Ø ˝”–√˜œ‘µƒÃÿµ„°£÷⁄À˘÷‹÷™£¨‘⁄¥´Õ≥µ• ∆⁄µƒΩ Ù‘≠◊”º‰∂‘ ∆∫Ø ˝÷–Ωˆøº¬«∫À◊”º‰œ‡ª•◊˜”√°£º”»ÎµÁ◊”∂‘∫À◊”µƒπ±œ◊∫Û£¨Ω Ù‘≠◊”º‰∂‘ ∆◊˜”√«˙œflæÕ±‰≥…¡Ω≤ø∑÷[11]£∫”…“ª∏ˆ«ø≈≈≥‚÷––ƒº”“ª◊È÷Ω•À•ºıµƒFriedelœÓππ≥…£¨”…¥Àø…“‘“˝»Î“ª¿‡’Î∂‘◊ºæßÃÂœµµƒ ∆∫Ø ˝°£À¸µƒÃÿµ„ «÷–≥§≥Ã◊˜”√≤ª «µ•µ˜±‰ªØ∂¯ «¥Ê‘⁄∂‡∏ˆ’Òµ¥œÓ£¨’Òµ¥œÓª·‘⁄ ∆«˙œfl÷–≤˙…˙ƒ‹¡øº´÷µµ„£¨‘⁄Ω·ππ…œ±Ìœ÷Œ™≥ˆœ÷∂‡∏ˆ—«Œ»µƒ‘≠◊”Œª÷√£¨—«Œ»Œª÷√¡Ω≤‡µƒ ∆¿›‘⁄“ª∂®Ãıº˛œ¬ø…“‘”––ßµÿ ¯∏ø‘≠◊”£¨¥”∂¯±£≥÷◊ºæߺ∞∆‰Ω¸À∆œ‡ÃÂœµµƒÃÿ ‚º∏∫ŒΩ·ππ°£¡ÌÕ‚£¨’Òµ¥µƒ ∆∫Ø ˝«˙œfl“≤∞¸∫¨√˜»∑µƒŒÔ¿Ì“‚“£¨º¥‘⁄∏flŒ¨ø’º‰[12]µ±÷–£¨∆Ω––ø’º‰(º¥ µø’º‰)µƒø’º‰±‰ªØ∂‘”¶”⁄…˘◊”£¨¥π÷±ø’º‰µƒø’º‰±‰ªØ∂‘”¶”⁄œ‡Œª◊”°£œ‡Œª◊”‘⁄ŒÔ¿Ìø’º‰µƒÃÂœ÷Œ™◊ºæß÷–ƒ≥–©‘≠◊”¥”‘≠¿¥µƒ—«Œ»Œª÷√∑≠π˝ ∆¿›Ã¯‘æµΩ∆‰Ω¸¡⁄µƒ¡Ì“ª—«Œ»Œª÷√[13]£¨‘⁄◊ºæßΩ·ππ÷–±Ìœ÷Œ™∆¥øÈ¡⁄Ω¸∂•µ„µƒ‘≠◊”º‰Ã¯‘æ°£
2 ◊ºæßΩ·ππº∆À„÷–µƒ≥£”√ ∆∫Ø ˝
2.1 ª˘”⁄ºÚµ•∂‘ ∆µƒ ∆∫Ø ˝
2.1.1 Dzugutov∂‘ ∆
Dzugutov∂‘ ∆[14] «µ•‘≠◊”ÃÂœµ÷–≥£”√µƒ“ª÷÷ ∆, À¸µƒÃ·≥ˆ «Œ™¡ÀΩ‚æˆ”…“∫èªÚ∏flŒ¬Ω Ù≤£¡ßŒ™≥ı ºÃ¨µƒÃÂœµ÷Ω•ÕÀªπ˝≥õ±÷–Ω·ππ‘≠◊”æ÷”ÚΩ·ππµƒŒ £¨∆‰±Ì¥Ô Ω»Á Ω(1)À˘ æ°£Dzugutov∂‘ ∆«˙œflµƒœ‘÷¯Ãÿµ„ «¥´Õ≥µƒLennard-Jones ∆«∞∞Î≤ø∑÷œ‡Õ¨£¨∂¯‘⁄ ∆ƒ‹◊Ó–°÷µ”ÎΩÿ∂œæ‡¿Î÷ƺ‰º”»Î“ª∏ˆ”–œfi∏flµƒ ∆¿›°£
¶µ(r)=¶µ1+¶µ2 (1)
∆‰÷–£∫
°›
£º
¶µ1=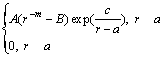
°›
£º
¶µ2=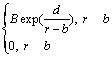
Ω÷–£∫r «‘≠◊”º‰æ‡¿Î£ªa∫Õc∑÷± «µ⁄1Ãı ∆«˙œflµƒΩÿŒ≤懿ΔΌ»∂®Œª÷√£ªb∫Õd «µ⁄2Ãı«˙œfl÷–a”Îc∂‘”¶µƒ≤Œ ˝£ªA£¨B∫ÕmŒ™µ˜’˚≤Œ ˝°£±Ì1¡–≥ˆ“ª◊ȃ£–Õµƒ ∆≤Œ ˝°£
±Ì1 Dzugutov∂‘ ∆∂‘”¶µƒ≤Œ ˝[14]
Table 1 Parameters of Dzugutov pair potential[14]
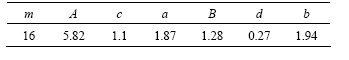
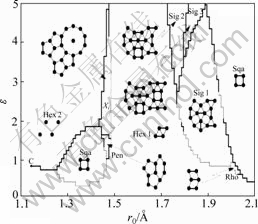
Õº1À˘ 挙±Ì1∂‘”¶µƒDzugutov∂‘ ∆µƒ ∆ƒ‹«˙œflÕº°£¥”Õº1ø…÷™£∫’˚∏ˆ ∆ƒ‹∞¸∫¨2∏ˆ ∆ƒ‹º´–°÷µ∫Õ1∏ˆº´¥Û÷µ£¨”…1∏ˆ«ø≈≈≥‚÷––ƒº”1∏ˆFriedelœÓππ≥…°£
”Î¥´Õ≥µƒ ∆∫Ø ˝“◊–Œ≥…√‹≈≈Ω·ππµƒÃÿ–‘≤ªÕ¨£¨Dzugutov ∆∏¸«˜œÚ”⁄–Œ≥…æ€Àƒ√ÊÃÂÕ≈¥ÿ°£Ã·≥ˆDzugutov ∆µƒ≥ˆ∑¢µ„ «‘⁄≤£¡ßè÷–µƒ”¶”√£¨«“À¸‘⁄’‚

Õº1 Dzugutov∂‘ ∆«˙œfl[14]
Fig.1 Curves of Dzugutov pair potential[14]
÷÷ÃÂœµµ±÷–µƒ”¶”√»°µ√¡À±»Ωœ∫√µƒΩ·π˚°£»Á“∫Ã¨Ω ÙªÚΩ Ù≤£¡ß‘⁄ÕÀª¿‰»¥π˝≥Ã÷–µ±¿‰»¥ÀŸ∂»◊„πª¬˝ ±ø…“‘–Œ≥…Œ»∂®µƒ Æ∂˛¥Œ◊ºæßΩ·ππ[15]°£πÿ”⁄Ω·ππ’Òµ¥–‘÷ µƒ—–æøø…“‘÷§√˜£¨¶ƒœ‡æßà«∂˛ Æ√ÊÃÂΩ Ù≤£¡ßÃÂœµµƒ∂‘”¶æßÃÂœ‡[16-17]°£MATTILAµ»[18]≤…”√Dzugutov ∆∫ÕEAM ∆∂‘∏flƒ‹¡£◊”“˝∑¢µƒ∑¯’’À…À∂‘Ni°¢Pµ•÷ º∞NiP≤£¡ßÃ¨Ω·ππÀÊ ±º‰µƒ”∞œÏΩ¯––¡Àƒ£ƒ‚£¨∑¢œ÷∞¸∫¨∂˛ Æ√ÊÖڵƒΩ·ππ‘⁄∆Ω∫‚◊¥Ã¨œ¬”–∫‹«øµƒπÃ∂®ƒ‹¡¶[19]°£¡ÌÕ‚£¨ π”√Dzugutov ∆∑¢œ÷Õÿ∆ÀΩ·ππµƒ≤ªÕ¨ª·µº÷¬≤ªÕ¨µƒ’Ò∂غ§∑¢∑Ω Ω£¨≤¢”Î∆‰≥€‘•Œ»∂®µƒΩ Ùœ‡’Ò∂Ø∑Ω ΩΩ¸À∆[20]°£
2.1.2 Lennard-Jones-Guass ∆(LJG)
”…”⁄Dzugutov ∆ «“ª÷÷’Î∂‘–‘µƒœ‡ª•◊˜”√ ∆£¨æ≠≥£”¶”√”⁄ºÚµ•µƒµ•≥…∑÷Ω Ù≤£¡ßÃÂœµ£¨œ‡∂‘∞¸∫¨µÁ◊”ƒ‹¡øøº¬«µƒ∂‘ ∆œ‡ª•◊˜”√£¨∏√ ∆∫Ø ˝ΩˆΩÿ∂œ‘⁄µ⁄“ª∏ˆFriedelœÓµƒº´–°÷µ¥¶£¨∂¯Ω´∆‰À˚÷–≥§≥ÃœÓ∫ˆ¬‘£¨“Ú∂¯£¨÷–≥§≥Ã◊˜”√±Ì’˜ƒ‹¡¶≤ª«ø°£“ª÷÷”––ß∑Ω∑® «‘⁄ ∆«˙œfl÷–—”≥§FriedelœÓµƒΩÿ∂œŒª÷√£¨LJG ∆[21]æÕ «∆‰÷–µƒ“ª÷÷£¨∆‰ ∆∫Ø ˝µƒ–Œ Ω»Áœ¬£∫
 (2)
(2)
LJG ∆∞¸∫¨6∏ˆ≤Œ ˝£¨ Ω(2)÷–«∞¡ΩœÓ–Œ≥…“ª∏ˆŒª”⁄r=d¥¶…Ó∂»¶≈µƒLennard-Jones«˙œfl£¨∂¯∫Ûº”»Î“ª∏ˆ∏flÀπœÓ£¨¶≈0”√”⁄µ˜’˚µ⁄∂˛ ∆ƒ‹◊ÓµÕ÷µ¥¶µƒ ∆⁄Â…Ó∂»£¨’‚∏ˆ⁄ ∆Œª”⁄r=r0¥¶£¨øÌ∂»Œ™d°£Õº2À˘ 挙“ª◊È∂‘”¶≤ªÕ¨r0µƒLJG ∆«˙œfl°£
LJG ∆µƒ‘≠¿Ì «‘⁄¥´Õ≥Lennard-Jones ∆µƒª˘¥°…œ‘⁄ ∆ƒ‹◊Ó–°÷µŒª÷√”ÎΩÿ∂œæ‡¿Î÷ƺ‰º”»Î“ª∏ˆ∏flÀπ∑÷≤º£¨”ÎDzugutov ∆œ‡±»£¨LJGµƒ“‚“Âœ‡µ±”⁄‘⁄«∞¡Ω∏ˆFriedel’Òµ¥œÓ÷ƺ‰Ω¯––Ωÿ∂œ£¨¥”∂¯µ√µΩ“ª∏ˆæfl”–∞¸∫¨À´ ∆⁄Â∫Õµ• ∆¿›µƒ◊˜”√∫Ø ˝£¨’‚—˘µƒΩ·π˚ «÷±Ω”“˝»Î“ª∏ˆŒ»∂®Œª÷√∫Õ“ª∏ˆ—«Œ»∂®Œª÷√°£∏˘æ›º´÷µµƒŒª÷√”Î ∆⁄Â…Ó∂»µƒ≤ªÕ¨£¨ø…“‘µ√µΩ≤ªÕ¨ª˘Ã¨µƒLJG∫Ø ˝°£
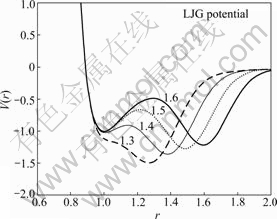
Õº2 ≤ªÕ¨r0 ±LJG ∆«˙œfl[22]
Fig.2 LJG potential for different r0[22]
ROTHµ»[22] π”√LJG ∆—–æø¡À∂˛Œ¨°¢»˝Œ¨Ω·ππœ¬Penrose∆¥øÈ≤ªÕ¨◊”Ω·ππ∏Òµ„µƒŒ»∂®–‘£¨∑¢œ÷∏Òµ„¥¶≤ªÕ¨µƒ◊”Õ≈¥ÿ∂—∆ˆÃÂœµŒ»∂®–‘”…◊”Õ≈¥ÿ±æ…̵ƒ–‘÷ æˆ∂®°£ENGEL∫ÕTREBIN[23]≤…”√∂˛Œ¨µ•‘≠◊”◊ºæßÃÂœµ£¨÷§√˜ Æ¥Œ◊ºæß «≤ª”…∆•≈‰πÊ‘Úøÿ÷∆µƒ¿ÌœÎÀʪ˙∆¥øÈΩ·π𣨑⁄¡ŸΩÁŒ¬∂»∏ΩΩ¸≥ˆœ÷µƒ◊ºæß”Î∆‰Ω¸À∆œ‡ø…ƒÊ◊™±‰ «”…¥Û¡ø∆¥øȃ⁄‘≠◊”ï‘æ µœ÷µƒ°£
LJG ∆◊Ó¥Ûµƒ”≈µ„‘⁄”⁄£¨À¸—–æøºÚµ•◊ºæßÃÂœµµƒΩ·ππ∫Õ»»¡¶—ß–‘÷ ø…“‘µ√µΩ∫‹∫√µƒΩ·π˚£¨∂¯«“À¸ƒ‹πª‘⁄ƒ£ƒ‚π˝≥õ±÷–÷±Ω”π€≤ϵΩ◊ºæßΩ·ππ”Î∆‰æßÃÂΩ¸À∆œ‡÷ƺ‰µƒœ‡ª•◊™ªØ°£Õº3À˘ 挙”…MC∑Ω∑®—–æøµ•‘≠◊”∂˛‘™ÃÂœµ÷–º”»»”ÎÕÀªπ˝≥Ã÷– Æ¥Œ∂‘≥∆∆¥øÈD”Îœ‡Œª◊””¶±‰¶÷1∫Õ¶÷2µƒ±‰ªØπʬ…£¨ø…“‘µ√µΩD∆¥øÈ(D tiles)±‰ªØπ˝≥à «ÕÍ»´ø…ƒÊµƒ°£∆‰÷–£∫Dec∫ÕXi∑÷±±Ì æ Æ¥Œ◊ºæß”Î∆‰∂‘”¶µƒΩ¸À∆æßÃÂœ‡£ªTm±Ì æÃÂœµµƒ»€µ„°£µ±T£æ0.4Tm ±£¨’˚∏ˆÃÂœµµ±÷–¶÷1∫Õ¶÷2»´≤øΩµŒ™0£¨±Ì√˜ÃÂœµ±‰Œ™ Æ¥Œ◊ºæߣ¨ÃÂœµ∑¢…˙œ‡±‰£¨‘⁄ÕÀªπ˝≥Ã÷–’˚∏ˆπ˝≥Ãø…ƒÊ°£
”ÎLennard-Jones ∆≤ªÕ¨£¨µ˜Ω⁄FriedelœÓŒª÷√ ∆⁄µƒŒª÷√∫Õ…Ó∂»£¨∂‘ÃÂœµÀ˘µ√µƒª˘Ã¨Ω·ππ¿‡–Õ≤˙…˙√˜œ‘µƒ”∞œÏ°£¥”Õº4ø…“‘µ√µΩ‘⁄Õ¨“ªµ•‘≠◊”ÃÂœµ÷–r0∫Õ…Ó∂»(¶≈)µƒ±‰ªØ£ª”…”⁄ ∆ƒ‹«˙œfl¥Ê‘⁄À´ ∆⁄£¨“Ú¥À£¨∫¨”–2∏ˆ ∆ƒ‹º´–°÷µµ„£¨œ‡µ±”⁄ÃÂœµµ±÷–≥ˆœ÷¡ΩÃ◊æß∏Ò≥£ ˝µƒπ≤Õ¨◊˜”√£¨¥”∂¯–Œ≥…≤ªÕ¨ª˘Ã¨Ω·ππ°£
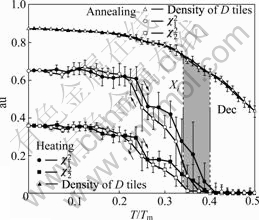
Õº3 Æ¥ŒÀʪ˙∆¥øÈ◊ºæß”Î∆‰Ω¸À∆œ‡µƒœ‡±‰[22]
Fig.3 Phase transition between decagonal RT and approximant ¶÷i[22]
Õº4 T=0, ¶ƒ2=0.02 ±LJG ∆Ω·ππœ‡Õº[22]
Fig.4 Phase diagram of LJG potential at T=0 and ¶ƒ2=0.02[22]
ENGEL∫ÕTREBIN[23]¥”µ•‘≠◊”ÃÂœµ÷–∑¢œ÷”…“∫èÃÂœµµ√µΩµƒ∂‡÷÷æß㨓‘º∞ Æ¥Œ◊ºæß∫Õ Æ∂˛¥Œ◊ºæß «ø…“‘Õ®π˝‘≠◊”◊‘◊È◊∞µ√µΩµƒ°£‘⁄ÕÀªπ˝≥Ã÷–£¨ÏÿŒ»∂®µƒ Æ¥Œ◊ºæßÕ®π˝œ‡Œª◊”ï‘æ µœ÷Ω·ππ÷ÿ≈≈œÚΩ¸À∆œ‡∑¢…˙ø…ƒÊµƒœ‡±‰£¨’˚∏ˆÃÂœµµƒ±‰ªØπ˝≥ñ̜÷Œ™≤ªÕ¨æß∏Ò≥£ ˝÷ƺ‰µƒæ∫’˘°£LJG ∆∂‘∑«æßΩ·ππÕ¨—˘æfl”–∫‹∫√µƒ√Ë ˆƒ‹¡¶£¨HOANG∫ÕODAGAKI [25]¥”µ•‘≠◊”ÃÂœµ“∫ÿ‰»¥µ√µΩµƒŒfi–ÚΩ·ππ≥ˆ∑¢£¨—–æø¡Àƒ…√◊ø≈¡£µƒΩ·ππ–‘÷ £¨≤¢∑¢œ÷¥Û¡øµƒ∂˛ Æ√ÊÃÂ∂Ã≥Öڰ£
2.1.3 –°Ω·
Dzugutov«„œÚ”⁄–Œ≥…æ€Àƒ√ÊÃÂΩ·π𣨒‚÷÷Ω·π𱻥´Õ≥∂‘ ∆◊˜”√ƒ‹πª∏¸”––ßµÿ±£≥÷—«Œ»∂®∏flƒ‹Ω·π𣨒‚÷÷Ω·ππÃÿ’˜∂‘”⁄◊ºæß–‘÷ ∑«≥£÷ÿ“™°£¿˚”√LJG ∆‘⁄∂˛Œ¨ÃÂœµµƒµ•¿‡‘≠◊”Ω·ππ÷–ƒ‹πªƒ£ƒ‚≥ˆ◊ºæßÔÎæßÃÂœ‡µƒø…ƒÊ–‘œ‡±‰°£Dzugutov ∆÷˜“™”√”⁄√Ë ˆ∂Ã≥Ãœ‡ª•◊˜”√£¨∂¯”√”⁄√Ë ˆ≥§≥Ã◊˜”√ ±”–√˜œ‘»±œ›£¨“Ú¥À£¨Œfi∑®µ√µΩ¥ø¥‚µƒ“∫èªÚ∆¯Ã¨Ω·ππÃÂœµ°£LJG ∆÷–µ⁄∂˛ ∆⁄µƒŒª÷√º∞∆‰…Ó∂»∂‘◊Ó÷’Ω·π𵃔∞œÏ“≤”¶µ±Ãÿ±◊¢“‚°£¡ÌÕ‚£¨À‰»ª¡Ω÷÷ ∆∂º»°µ√¡À∫‹∫√µƒΩ·π˚£¨µ´ «À¸√«»‘»ª «“ª÷÷æ≠—È–‘ ∆∫Ø ˝£¨—–æø∂‘œÛœ‡∂‘ºÚµ•£¨‘⁄Ω‚æˆ∂‡‘™◊ºæß”ÎΩ¸À∆œ‡Œ Â…œ»‘»ª¥Ê‘⁄Ωœ¥Û≤ª◊„°£
2.2 Realistic–Õ ∆∫Ø ˝
2.2.1 Moriarty-Widom”––ß∂‘ ∆
Œ™¡ÀΩ‚æˆ∂‡‘™ÃÂœµ÷–◊ºæßΩ·ππŒ £¨∑¢’π¡Àæ´∂»∏¸∏flµƒRealistic–Õ ∆∫Ø ˝(Realistic potentials)°£’‚÷÷ ∆“‘µ⁄“ª–‘‘≠¿ÌªÚ µ—ÈΩ·π˚Œ™ª˘¥°£¨Ωœ÷Æ«∞µƒ ∆∫Ø ˝◊º»∑∂»”–¡À∫‹¥Û÷∏fl°£
MORIARTY∫ÕWIDOMµ»[26-27]¥”√‹∂»∑∫∫ؿ̬€≥ˆ∑¢£¨Ω´µ⁄“ª–‘‘≠¿Ì÷–µƒDFTº∆À„Ω¯––º∂ ˝’πø™£¨≤¢≤…”√œ‡πÿµƒŒÔ¿ÌΩ¸À∆£¨∏¯≥ˆ∏¥‘”∫œΩÃÂœµ÷–Al”Îπ˝∂…Ω Ùœ‡ª•◊˜”√µƒ”––ß∂‘ ∆◊˜”√°£Õº5À˘ 挙»˝‘™◊ºæßÃÂœµAlNiCo÷–µƒMoriarty-Widom”––ß∂‘ ∆£¨¥”∂‘ ∆«˙œflø…“‘ø¥≥ˆ£¨√ø÷÷∂‘ ∆◊˜”√∂º∞¸∫¨√˜œ‘µƒ’Òµ¥––Œ™£¨’‚”ο̬€÷–µƒFriedel’Òµ¥“ª÷¬£¨Õ¨ ±¥Û¡ø—«Œ»∂®Œª÷√µƒ¥Ê‘⁄ƒ‹πª”––ßµÿŒ¨≥÷ÃÂœµΩ·ππµƒœ‡∂‘Œ»Ã¨°£
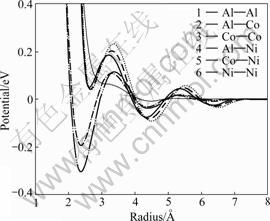
Õº5 AlNiCoÃÂœµ÷–Moriarty-Widom”––ß∂‘ ∆[26]
Fig.5 Moriarty-Widom effective pair potentials for decagonal AlNiCo[26]
MIHALKOVI?µ»[28]“‘Àʪ˙∆¥øȵƒ36?∫Õ72?’‚2÷÷¡‚–ŒΩ·ππŒ™ª˘¥°£¨¿˚”√ µ—ȵ√µΩµƒ◊ºæßÃÂœµµƒ–‘÷ ”ÎMoriarty-Widom”––ß∂‘ ∆£¨Õ®π˝Monte Carloƒ£ƒ‚∑¢œ÷£¨‘⁄œµÕ≥ƒ‹¡ø◊ÓµÕµƒÃıº˛œ¬ø…“‘µ√µΩ∆¥øÈ√Ë ˆµƒ◊Ó–°œfi÷∆(Minimally constrained)”Î∏flœfi÷∆(Highly constrained simulation)2÷÷Ãıº˛µƒ◊ºæßΩ·ππ°£Ω·π˚∑¢œ÷£¨µ√µΩµƒÃÂœµΩ·ππ”Î µ—ȵ√µΩµƒΩ·ππœ‡±»ΩˆΩˆ «ÕÍ’˚µƒ Ʊfl–Œµƒ±Ìπ€√‹∂»∆´µÕ£¨’‚ø…ƒ‹ «”…”⁄’Ê µΩ·ππ÷–µƒœ‡Œª◊”∂—∂‚Œfi–Úµº÷¬ Ʊfl–Œµƒ±Ìπ€√‹∂»…œ…˝[29]°£BOISSIEUµ»[30]¿˚”√∏√∂‘ ∆∫‹∫√µÿ÷ÿœ÷¡À¥” µ—È÷–µ√µΩµƒZn-Mg-Sc◊ºæßÃÂœµ”Î∆‰Zn-ScæßÃÂœ‡µƒ∫·œÚ…´…¢πÿœµ”ÎΩ·ππ“Ú◊”Ãÿ’˜£¨À¸‘⁄Al-Ni-CoÃÂœµµƒ–‘÷ ”ÎΩ·ππº∆À„…œ“≤µ√µΩ¡À÷ÿ“™µƒΩ·π˚[31-33]°£
HOCKER∫ÕG?HLER[34] π”√Moriarty-Widom∂‘ ∆—–æø¡ÀAlNiCo”ÎAlCuCoœ‡πÿÃÂœµ÷–Al‘≠◊”¿©…¢––Œ™£¨∑¢œ÷ÃÂœµ÷–Õ¨÷÷‘™Àÿ‘⁄≤ªÕ¨‘≠◊”ª∑æ≥œ¬ªÓ∂Ø–‘≤Ó“Ï√˜œ‘£¨‘⁄T£æ0.6 Tm ±∑¢…˙«ø¡“µƒAl‘≠◊”¿©…¢œ÷œÛ°£Õº6∏¯≥ˆ¡À4÷÷ Æ¥Œ◊ºæßΩ·ππ÷–Al‘≠◊”¿©…¢µƒArrheniusπÿœµ°£∆‰÷–£∫xŒ™◊º÷‹∆⁄–‘∑ΩœÚ£ªzŒ™÷‹∆⁄–‘¿©…¢∑ΩœÚ°£¥”Õº6ø…÷™‘⁄4÷÷Ω·ππ÷–Al‘⁄÷‹∆⁄–‘∑ΩœÚµƒ¿©…¢ÀŸ∂»√˜œ‘±»◊º÷‹∆⁄∑ΩœÚµƒøÏ£¨∂¯«“æfl”–±»÷‹∆⁄–‘∑ΩœÚ∏¸¥Ûµƒ¿©…¢º§ªÓƒ‹£¨±Ì æAl‘≠◊”∏¸»›“◊‘⁄÷‹∆⁄–‘∑ΩœÚ∑¢…˙¿©…¢°£

Õº6 AlCuCoÃÂœµ÷–Al‘≠◊”¿©…¢µƒArrheniusπÿœµ[31]
Fig.6 Arrhenius plots for Al diffusion in AlCuCo[31]
2.2.2 ¡¶∆•≈‰∑Ω∑® ∆
Œ™¡ÀΩ‚æˆ“ª∞„–‘µƒÃÂœµœ‡ª•◊˜”√£¨ ERCOLESSI∫ÕADAMS[35]÷≥ˆ¡À°∞¡¶∆•≈‰∑Ω∑®(Force-match method)°±£¨À¸ «Õ®π˝∏¯∂®ÃÂœµ–Ë“™øº¬«µƒ≤ŒøºΩ·π𣨗°»°ƒ£–Õ ∆µƒ¿‡–Õ£¨¿˚”√µ⁄“ª–‘‘≠¿Ìµ√µΩπÿ”⁄¡¶°¢”¶¡¶∫Õƒ‹¡øµ»∑Ω√Êœ‡∂‘◊º»∑µƒ ˝æ›£¨»ª∫Û£¨¿˚”√”≈ªØµƒ∑Ω∑® π”…ƒ£–Õ ∆µ√µΩµƒ∂‘”¶¡ø≤ª∂œ±∆Ω¸µ⁄“ª–‘‘≠¿Ì÷µ£¨ π ∆∫Ø ˝æ´∂»µ√µΩ÷∏fl°£ ∆≤Œ ˝Õ®π˝°∞¡¶∆•≈‰∑Ω∑®°±”… Ω(3)ªÒµ√°£
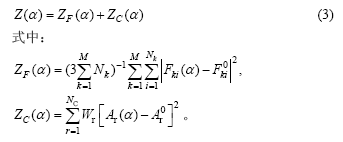

ÀÊ◊≈Z÷µ ’¡≤º∆À„÷µ÷Ω•±∆Ω¸µ√µΩµ⁄“ª–‘‘≠¿Ì÷µµƒΩ¸À∆÷µ£¨≤¢”…¥Àµ√µΩ ∆≤Œ ˝¡–¶¡µƒ”≈ªØ÷µ°£
µ±«∞ π”√°∞¡¶∆•≈‰∑Ω∑®°±µ√µΩµƒ∏¥‘”∫œΩœ‡÷–µƒ‘≠◊”º‰œ‡ª•◊˜”√ ∆“—æ≠µ√µΩ¡À∫‹∫√µƒ—È÷§£¨Õ®π˝∂‘∂˛Œ¨◊º÷‹∆⁄µƒAlNiCo≤„◊¥Ω·ππ¡¨–¯º”Œ¬µƒ∑Ω∑®µ√µΩ”ÎMoriarty-Widom∂‘ ∆Ω·π˚Œ«∫œµƒÃÂœµ»»Œ»∂®–‘£¨‘⁄0.5Tmœ¬ÃÂœµΩ·ππŒfi√˜œ‘±‰ªØ£¨œ‡Œª◊”‘æ«®ª˙¬ ∫‹–°£¨‘≠◊”‘À∂Ø∑ΩœÚΩˆΩˆºØ÷–‘⁄÷‹∆⁄∑ΩœÚµƒ‘≠◊”√Ê÷ƃ⁄£¨≤¢µ√µΩ¡À”Î µ—ÈΩ·π˚œ‡Ω¸µƒ»€µ„°£Õ®π˝’‚÷÷ ∆º∆À„¡ÀAlNiCoÃÂœµ∏flŒ¬œ¬µƒAl‘≠◊”¿©…¢£¨µ√µΩ¡À∫‹∫√µƒΩ·π˚[36]°£¡ÌÕ‚£¨À¸‘⁄∞¸∫¨∂˛ Æ√ÊÃÂΩ·ππµƒ∂˛ Æ√ÊÃÂ◊ºæßΩ·ππ÷–“≤µ√µΩ¡À∫‹∫√µƒ”¶”√£¨‘⁄—–æø∂˛ Æ√ÊÃÂCaCd◊ºæßΩ·ππµƒ1/1◊ºæßΩ¸À∆œ‡CaCd6ππµƒŒfi–ÚªØ◊™±‰ π˝≥Ã÷–£¨µ√µΩ”Î µ—È÷µ∑«≥£Ω”Ω¸µƒŒfi–ÚªØ◊™±‰Œ¬ ∂»[37-39]°£≥˝¡À…œ ˆΩ·π˚£¨¡¶∆•≈‰∑Ω∑®ªπ‘⁄NbCr2÷–µƒ¡—Œ∆¿©’π[40]”ÎZn2Mgµƒ∂Ø¡¶—ßΩ·ππ“Ú ˝[41]µƒº∆À„…œ»°µ√¡À∫√µƒΩ·π˚°£
2.2.3 –°Ω·
Moriarty-Widom∂‘ ∆≤…”√º∂ ˝’πø™∑Ω∑®µ√µΩ¡À∞¸∫¨¿‡FriedelœÓµƒ∂‡º∂’Òµ¥¡Ωú‰œ‡ª•◊˜”√°£”ÎMoriarty-Widom∂‘ ∆πÃ∂®µƒ±Ì¥Ô–Œ Ω≤ªÕ¨£¨¡¶∆•≈‰∑Ω∑®µ√µΩµƒ ∆µƒ±Ì¥Ô–Œ Ω±»Ωœ¡ÈªÓ£¨ ∆∫Ø ˝–Œ Ω÷˜“™»°æˆ”⁄—°»°µƒƒ£–Õ ∆∫Ø ˝£¨Õ®π˝µ⁄“ª–‘‘≠¿Ì÷µ∂‘≤Œøº÷µΩ¯––µ¸¥˙–fi’˝°£’‚¡Ω÷÷ ∆∂º «ª˘”⁄µ⁄“ª–‘‘≠¿Ì÷µ£¨ƒ‹πª”––߃£ƒ‚∂‡‘™◊ºæß”Î∆‰Ω¸À∆œ‡µƒΩ·ππ∫Õ–‘÷ °£”…”⁄¡¶∆•≈‰∑Ω∑®–Œ Ω¡ÈªÓ£¨ø…“‘”¶”√EAMµ» ∆ƒ£–Õ£¨ π”√∑Ω±„£¨∆‰÷˜“™”≈µ„‘⁄”⁄Ω·∫œ¡À¥´Õ≥ ∆µƒº∆À„ƒ‹¡¶∫Õµ⁄“ª–‘‘≠¿Ìµƒ◊º»∑–‘£¨ πµ√œ‡ª•◊˜”√ ∆µƒæ´∂»”–¡À∫‹¥Û÷∏fl°£µ´ ∆µƒ◊˜”√∑∂Œßæ÷œfi”⁄ ∆µ˜Ω⁄π˝≥Ã÷–µƒº∏÷÷Ãÿ ‚Ω·ππ£¨∂¯«“ ∆ƒ‚∫œπ˝≥õ±÷–µƒπ˝∂‡±‰¡ø π ∆∫Ø ˝µ˜Ω⁄±»Ωœ¿ßƒ—°£
3 ◊‹Ω·”Î’πÕ˚
1) ∂‘◊ºæßΩ·π𵃠∆∫Ø ˝Ãÿµ„º∞≥£”√ ∆∫Ø ˝Ω¯––¡ÀΩœŒ™œµÕ≥µƒ◊‹Ω·£¨∑¢œ÷∂‘ ∆œÓ÷–º”»ÎµÁ◊”∂‘∫À◊”◊˜”√∫Û£¨◊ºæßΩ·ππ÷–‘≠◊”º‰≥§≥Ã◊˜”√ø…ƒ‹≤¢≤ª «µ•µ˜±‰ªØµƒ∂¯ «¥Ê‘⁄∂‡∏ˆ’Òµ¥œÓ£¨À¸ƒ‹”––ߌ¨≥÷◊ºæßÃÿ ‚µƒº∏∫ŒΩ·ππÃÿ’˜£¨Õ¨ ±’Òµ¥œÓ‘⁄ ∆«˙œfl÷–≤˙…˙»Ù∏…∏ˆƒ‹¡øº´÷µµ„£¨”Î◊ºæßÃÂœµµƒΩœ∏flƒ‹¡øœ‡∂‘”¶£¨“Ú¥À£¨√Ë ˆ◊ºæßΩ·ππ ±≤…”√µƒ ∆∫Ø ˝«˙œfl”Î√Ë ˆæßè∫Õ∑«æßè ±”–À˘≤ªÕ¨°£
2) ÀÊ◊≈œ÷¥˙º∆À„ª˙»Ì”≤º˛µƒ∑¢’π£¨æ´∂»Ωœ∏flµƒµ⁄“ª–‘‘≠¿Ìº∆À„ƒ‹πª¥¶¿Ì‘Ω¿¥‘Ω¥Ûµƒ‘≠◊”ÃÂœµ∫Õº∆À„‘Ω¿¥‘Ω∂‡µƒ≤ƒ¡œ–‘÷ £¨µ´ «∂‘”⁄¥Û∂¯∏¥‘”µƒ◊ºæßÃÂœµ»‘»ªŒfi∑®¥¶¿Ì°£Õ®π˝”ε⁄“ª–‘‘≠¿Ìº∆À„∫Õ µ—È≤‚¡øΩ·π˚œ‡Ω·∫œ£¨ƒ‚∫œµ√µΩ”–πÿæ≠—È ∆∫Ø ˝≤Œ ˝£¨ «“ª÷÷∂‘◊ºæßΩ·ππ∫Õ–‘÷ Ω¯––‘≠◊”≥fl∂»ƒ£ƒ‚µƒ”––ß∑Ω∑®°£
REFERENCES
[1] LENNARD-JONES J E. On the determination of molecular fields [J]. Proc Roy Soc A, 1924, 106: 463-469.
[2] TERSOFF J. New empirical model for the structural properties of silicon [J]. Phys Rev Lett, 1986, 56: 632-635.
[3] DAW M S, BASKES M I. Semiempirical, quantum mechanical calculation of hydrogen embrittlement in metals [J]. Phys Rev Lett, 1983, 50: 1285-1288.
[4] DAW M S, BASKES M I. Embedded atom method: Derivation and application to impurities, surface and other defects in metals [J]. Phys Rev B, 1984, 29: 6443-6453.
[5] ≈∑—Ù“Â∑º, ÷”œƒ∆Ω. ƒ˝æ€Ã¨ŒÔ÷ º∆À„∫Õƒ£ƒ‚÷– π”√µƒœ‡ª•◊˜”√ ∆[J]. ¡¶—ßΩ¯’π, 2006, 36(3): 321-343.
OUYANG Yi-fang, ZHONG Xia-ping. Interatomic potentials of computer simulation of condensed matters [J]. Advances In Mechanics, 2006, 36(3): 321-343.
[6] ’≈∞ÓŒ¨, ∫˙Õ˚”Ó, Ê–°¡÷. «∂»Î‘≠◊”∑Ω∑®¿Ì¬€º∞∆‰‘⁄≤ƒ¡œø∆—ß÷–µƒ”¶”√: ‘≠◊”≥fl∂»≤ƒ¡œ…˺∆¿Ì¬€[M]. ≥§…≥: ∫˛ƒœ¥Û—ß≥ˆ∞Ê…Á, 2003.
ZHANG Bang-wei, HU Wang-yu, SHU Xiao-lin. Theory of embedded atom method and its application to materials sscience: Atomic scale materials design theory [M]. Changsha: Hunan University Press, 2003.
[7] Õı‘¬ª™, ¡ı—fiœ¿, Õı —∑. Ω Ùº∞∫œΩ÷–µƒ‘≠◊”º‰œ‡ª•◊˜”√ ∆[J]. ¡…ƒ˛¥Û—ß—ß±®, 2006, 33(1): 24-28.
WANG Yue-hua, LIU Yan-xia, WANG Xun. Interatomic potentials in metal and alloy [J]. Journal of Liaoning University, 2006, 33(1): 24-28.
[8] ≥¬ «ø, ≤‹∫Ï∫Ï, ª∆∫£≤®. ∑÷◊”∂Ø¡¶—ß÷– ∆∫Ø ˝—–æø[J]. ÃÏΩÚ¿Ìπ§—ß‘∫—ß±®, 2004, 20(2): 101-105.
CHEN Qiang, CAO Hong-hong, HUANG Hai-bo. A research on the interatomic potential in molecular dynamics (MD) [J]. Journal of Tianjin Institute of Technology, 2004, 20(2): 101-105.
[9] LI J H, DAI X D, LIANG S H, TAI K P, KONG Y, LIU B X. Interatomic potentials of the binary transition metal systems and some applications in materials physics [J]. Physics Reports, 2008, 455: 1-134.
[10] ERKOC S. Empirical many°™Body potential energy functions used in computer simulations of condensed matter properties [J]. Phys Reports, 1997, 278: 79-105.
[11] HAFNER J. From Hamiltonians to phase diagrams [M]. Berlin: Springer-Verlag, 1987.
[12] DUNEAU M, KATZ A. Quasiperiodic patterns [J]. Phys Rev Lett, 1985, 54: 2688-2691.
[13] ÃÔ ∫◊. ∏¥‘”Ω·ππ∫œΩœ‡÷–Ãÿ ‚»±œ›µƒœ‘Œ¢Ω·ππ—–æø[D]. ±±æ©: ÷–π˙ø∆—ß‘∫ŒÔ¿Ì—–æøÀ˘, 2006: 31.
TIAN He. TEM study of microstructures of defects in the structurally complex metallic alloy phases [D]. Beijing: Institute of High Physics, Chinese Academy of Sciences, 2006: 31.
[14] DZUGUTOV M. Glass formation in a simple monatomic liquid with icosahedral inherent local order [J]. Phys Rev A, 1992, 46: R2984-R2987.
[15] DZUGUTOV M, SIMDYANKIN S I, ZETTERLING F H. Decoupling of diffusion from structural relaxation and spatial heterogeneity in a supercooled simple liquid [J]. Phys Rev Lett, 2002, 89: 195701-4.
[16] SIMDYANKIN S I, TARASKIN S N, DZUGUTOV M, ELLIOTT S R. Vibrational properties of the one-component ¶“ phase [J]. Phys Rev B, 2000, 62: 3223-3231.
[17] SIMDYANKIN S I, TARASKIN S N, ELENIUS M, ELLIOTT S R, DZUGUTOV M. Nature of vibrational eigen modes in topologically disordered solids [J]. Phys Rev B, 2002, 65: 104302-7.
[18] MATTILA T, NIEMINEN R M, DZUGUTOV M. Simulation of radiation-induced structural transformation in amorphous metals [J]. Phys Rev B, 1996, 53: 192-200.
[19] SADIGH B, DZUGUTOV M, ELLIOTT S R. Vacancy ordering and medium-range structure in a simple monatomic liquid [J]. Phy Rev B, 1999, 59: 1-4.
[20] SIMDYANKIN S I, DZUGUTOV M, TARASKIN S N, ELLIOTT S R. Connection between vibrational dynamics and topological order in simple glasses [J]. Phys Rev B, 2001, 63: 184301-5.
[21] STEIGERWALD M L, BRUS L E, ALIVISATOS A. Synthesis, stabilization, and electronic structure of quantum semiconductor nanoclusters [J]. Annu Rev Mater Sci, 1989, 19: 471-495.
[22] ROTH J, SCHILIING R, TREBIN H R. Stability of monatomic and diatomic quasicrystals and the influence of noise [J]. Phys Rev B, 1990, 41: 2735-2747.
[23] ENGEL M, TREBIN H R. Stability of the decagonal quasicrystal in the Lennard-Jones-Gauss system [J]. Phil Mag, 2008, 88: 1959-1965.
[24] ENGEL M, TREBIN H R. Self-assembly of monatomic complex crystals and quasicrystals with a double-well interaction potential [J]. Phys Rev Lett, 2007, 98: 225505-4.
[25] van HOANG V, ODAGAKI T. Molecular dynamics simulations of simple monatomic amorphous nanoparticles [J]. Phys Rev B, 2008, 77: 125434-11.
[26] MORIARTY J A, WIDOM M. First-principles interatomic potentials for transition-metal aluminides: Theory and trends across the 3d series [J]. Phys Rev B, 1997, 56: 7905-7917.
[27] WIDOM M, AL-LEHYANI I, MORIARTY J A. First-principles interatomic potentials for transition-metal aluminides (III). Extension to ternary phase diagrams [J]. Phys Rev B, 2000, 62: 3648-3657.
[28] MIHALKOVI? M, AL-LEHYANI I, COCKAYNE E, HENLEY C L, MOGHADAM N, MORIARTY J A, WANG Y, WIDOM M. Total-energy-based prediction of a quasicrystal structure [J]. Phys Rev B, 2002, 65: 104205-6.
[29] HENLEY C L, ELSER V, MIHALKOVI? M. Structure determinations for random-tiling quasicrystals [J]. Z Kristallogr, 2000, 215: 553-568.
[30] de BOISSIEU M, FRANCOUAL S, MIHALKOVI? M, et al. Lattice dynamics of the Zn-Mg-Sc icosahedral quasicrystal and its Zn-Sc periodic 1/1 appromant [J]. Nature Materials, 2007, 6: 977-984.
[31] JORAMATSU S, ISHII Y. Dynamic properties of Al-Ni-Co decagonal quasicrystals modeled by Ab initio interatomic pair potential [J]. J Phys Soc Jpan, 2007, 76: 034601-6.
[32] HIRAMATSU S, ISHII Y. Theoretical prediction of phase diagrams for Al-Co-Ni decagonal quasicrystals [J]. J Phys Soc Jpn, 2006, 75: 054602-6.
[33] GU N, HENLEY C L, MIHALKOVI? M. Co-rich decagonal Al-Co-Ni: Predicting structure, orientational order and puckering [J]. Phil Mag, 2006, 86: 593-599.
[34] HOCKER S, G?HLER F. Aluminium diffusion in decagonal quasicrystals [J]. Phys Rev Lett, 2004, 93: 075901-4.
[35] ERCOLESSI F, ADAMS J B. Interatomic potentials from first-principles calculations: The force-matching method [J]. Euro Phys Lett, 1994, 26: 583-588.
[36] HOCKER S, G?HLER F, BROMMER P. Molecular dynamics simulation of aluminium diffusion in decagonal quasicrystals [J]. Phil Mag, 2006, 86: 1051-1057.
[37] BROMMER P, G?HLER F, MIHALKOVI? M. Ordering and correlation of cluster orientations in CaCd6 [J]. Phil Mag, 2007, 87: 2671-2677.
[38] TAMURA R, EDAGAWA K, MURAO Y, TAKEUCHI S, SUZUKI K, ICHIHARA M, ISOBE M, UEDA Y. Order-disorder transition in cubic Cd6Yb and Cd6Ca [J]. J Non-cryst Solids, 2004, 334/335: 173-176.
[39] TAMURA R, NISHIMOTO K, TAKEUCHI S, EDAGAWA K, ISOBE M, UEDA Y. Universal low-temperature phase transition in Zn- and Cd-based crystalline approximants [J]. Phys Rev B, 2005, 71: 092203-4.
[40] R?SCH F, TREBIN H R, GUMBSCH P. Interatomic potentials and the simulation of fracture: C15NbCr2 [J]. Int J Fracture, 2006, 139: 517-526.
[41] BROMMER P, de BOISSIEU M, EUCHNER H, FRANCOUAL S, G?HLER F, JOHNSON M, PARLINSKI K, SCHMALZL K. Vibrational properties of MgZn2 [J]. Z Kristallogr, 2009, 224: 97-100.
(±‡º≠ —Ó ª™)
ª˘ΩœÓƒø£∫π˙º“◊‘»ªø∆—ߪ˘Ω◊ ÷˙œÓƒø(50671035, 50871038)
’∏»’∆⁄£∫2009-08-30£ª–fi∂©»’∆⁄£∫2009-11-30
Õ®–≈◊˜’fl£∫∫˙Õ˚”Ó£¨Ωà ⁄£¨≤© ø£ªµÁª∞£∫0731-88823971£ªE-mail: wangyuhu2001cn@yahoo.com.cn

