Theoretical analysis of β-LaNi5Hx (5≤x≤8): structure and energetics
来源期刊:中国有色金属学报(英文版)2005年第5期
论文作者:刘杨 吴锋
文章页码:1031 - 1039
Key words:LaNi5 hydride; geometry; electronic structure; heat of formation
Abstract: The calculations of total energy, band structure, and electronic density of states and Mulliken population analysis of β-LaNi5Hx(5≤x≤8) were performed by adopting the method of total energy based on the density functional theory. The augmented plane wave function was selected as the basis set in combination with ultra-soft pseudo-potential technology. The influence of the amount of H absorbed in alloys was discussed in terms of geometry, electronic structure and thermodynamic derived from calculated results. The results show that the amount of H absorbed and the preferred site occupation of the absorbed hydrogen atoms were controlled by the position of H-bands and the energy gap between H-bands and conduction bands. The β-phase hydrides of LaNi5 are most stable when hydrogen atom capacity coating in the range of 6-7.
Article ID: 1003-6326(2005)05-1031-09
LIU Yang(刘 杨), WU Feng(吴 锋)
(National Development Center of Hi-Tech Green Materials, Beijing Institute of Technology,Beijing 100081, China)
Abstract: The calculations of total energy, band structure, and electronic density of states and Mulliken population analysis of β-LaNi5Hx(5≤x≤8) were performed by adopting the method of total energy based on the density functional theory. The augmented plane wave function was selected as the basis set in combination with ultra-soft pseudo-potential technology. The influence of the amount of H absorbed in alloys was discussed in terms of geometry, electronic structure and thermodynamic derived from calculated results. The results show that the amount of H absorbed and the preferred site occupation of the absorbed hydrogen atoms were controlled by the position of H-bands and the energy gap between H-bands and conduction bands. The β-phase hydrides of LaNi5 are most stable when hydrogen atom capacity coating in the range of 6-7.
Key words: LaNi5 hydride; geometry; electronic structure; heat of formation CLC number: TG111; TP139+.7
Document code: A
1 INTRODUCTION
LaNi5-based alloys are the negative electrode material widely used in Ni/MH battery presently[1-3]. Many macroscopic prosperities of the material are related with its microscopic structure. It is necessary to properly describe the energy and electronic structure of LaNi5-based alloys and their hydrides. So far, much work on experiment[4-6] and theory[7-12] have been reported. Ultraviolet photoelectron spectroscopy (UPS) was used to measure the electronic specific heat coefficient[4-6] to obtain the electronic change on the Fermi surface before and after alloys hydrogenation. X-ray absorbed near edge spectroscopy(XANES)[7-9] was used to observe the nature of bonds between metal and hydrogen. The calculation method basing on the first principle theory, such as tight binding approximation recursion(TBR)[10], self-consistent linear muffin tin orbital(LMTO)[11] and DV-Xα[12], was adopted to analyse the electron density of LaNi5 and LaNi5H7. However, the location of the atoms in TBR is in the real space and the periodicity is not considered. And in the atomic sphere approximation of LMTO, the number of the atomic spheres influences the calculated results significantly. Additionally, DV-Xα is based on the cluster and takes only the nearest neighbor atoms into account, so it can hardly pay attention to the influence by farther atoms. In this work, the total energy-plane wave pseudopotential method was introduced with better description of large periodicity system and better analysis to the energy and electronic structure in the hydrogen interstice site in LaNi5.
2 DETAILS OF CALCULATIONS
2.1 Crystal structure
LaNi5 is the parent alloy in AB5-type hydrogen storage alloys with a hexagonal symmetry (space group P6/mmm, structure type CaCu5). La occupies the 1a (0, 0, 0) site and the two nonequivalent Ni occupy the 2c (1/3, 2/3, 0) and 3g (1/2, 0, 1/2) sites. Two ordered structures about its β-phase hydrides have been reported based on a careful interpretation of the neutron diffraction data. One is P6/mmm[13] with five nonequivalent interstices available for the hydrogen and their positions are given in Table 1. In the five sites, 3f is octahedron and others are tetrahedron. Another is P63mc[14, 15], which have a doubled unit cell of LaNi5 along the c-axis and a replacement of the six-fold symmetry axis by a 63 screw axis, with three different hydrogen sites (Table 1). In this work, both models were discussed and the initial crystal structures are listed in Table 2 and Table 3.
2.2 Calculation method
The total energy-plane wave pseudo-potential method[16] based on the density functional theory, applied for describing the large-scale periodic systems, was adopted. The total energy Eks as a function of electron density ρ(x) can be expressed by
Eks[ρ(x)]=Ts[ρ(x)]+Ees[ρ(x)]+
EXC[ρ(x)]+Eext[ρ(x)](1)
where the terms refer to the kinetic energy Ts of non-interacting electrons, the electrostatic energy Ees, the exchange-correlation energy EXC, and the potential energy Eext of non-interacting electrons in the external field.
Table 1 Interstitial sites in unit cell of LaNi5 (space group, P6/mmm) and La2Ni10 (space group, P63mc)
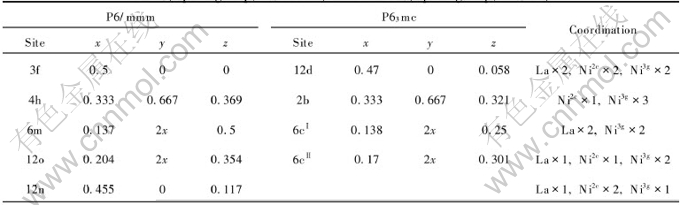
Table 2 Position of H in β-LaNi5Hx unit cell before and after geometry optimization
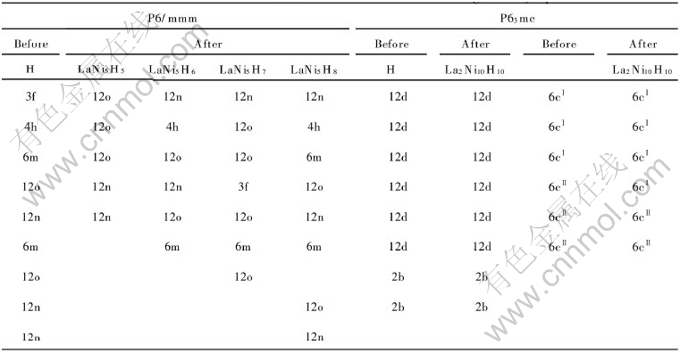
Table 3 Cell parameters of β-LaNi5Hx
The generalized gradient approximation (GGA)[17] combined in local density approximation (LDA) was used for the exchange-correlation energy functional. The ultra-soft pseudo-potential (USP)[18] describes the electron-ion interaction and generates good scattering properties over a pre-specified energy range, which results in much better transferability and accuracy of pseudo-potential. The pseudo-potential basis set in this work comprised a 5s25p65d16s2 valence configuration for La, a 3d84s2 valence configuration for Ni and a 1s1 valence configuration for H. The method for electronic relaxation is based on density mixing[19] combined with conjugate gradient to minimize the sum of eigenvalue. The method is robust for metallic systems for the cell elongated in one dimension. The solution of Kohn-Sham equation can be obtained by self-consistent. The new charge density at the end of the steps mixed with the initial density and the process is repeated until convergence.
In the calculation, the models were transformed to crystalline super-lattice firstly and then geometry optimization was performed on the models in the lowest symmetry (space group, P1). The single energy was calculated on the optimized models. In order to meet the convergence goal, the plane-wave cutoff energy was 240eV, and the FFT grid was 24×24×18 for LaNi5Hx(x=5-8) and 24×24×40 for La2Ni10H14. The calculation was performed in reciprocal space. When the self-consistent process ended, the energy change per atom was less than 0.2×10-4eV/atom, RMS displacement, force and stress was less than 10-4nm, 0.005eV/nm and 0.1GPa, respectively. The plane-wave cutoff energy in single point energy calculation was 300eV, FFT grid was 27×27×24 for LaNi5Hx(x=5-8) and 27×27×45 for La2Ni10-H14. The k-space is 0.0035nm. In the space, according Monkhorst-Pack algorithm, the Г-centered k-point grids in hexagonal irreducible Brilliouin zones (IBZ) was (6, 6, 7) with 126 k-points for LaNi5Hx(x=5-8) and (6, 6, 3) k-point grids with 98 k-points in IBZ for La2Ni10H14. The simulated environment temperature is 273K. Calculation of the H2 molecule was made using a supercell of 0.10×0.10×0.10nm3.
3 RESULTS AND DISCUSSION
3.1 Optimized structures
The cell parameters and the position of hydrogen after optimization are listed in Table 2 and Table 3. Three types of hydrogen motion occur in LaNi5Hx:
1) The tetrahedron sites 4h, 6m and 12o belong to the pentagon ring in a (110) plane[20]. Hydrogen atoms move to 12o site or stay at their original position. It is a fast diffusive motion that can easily occur around the z=1/2 plane, i.e. a hydrogen atom at 6m site move to the 12o site in LaNi5H5, LaNi5H6 and LaNi5H7, and hydrogen at 4h site to the 12o site in LaNi5H5 and LaNi5H7. It seems that the only way for a hydrogen atom to escape from a ring of 6m sites is to jump to a 12o site. A hydrogen atom in a 4h site can jump in a z direction to another 4h site, but then it must jump to a 12o site or simply return to its origin, which can be observed in the optimization process.
2) A rapid localized jump happens within the 3f-12n site clusters consisting of four 12n sites centered on a 3f site of the z=0 plane. It can be understood easily because the distances between nearest-neighbor sites are rather short (3f-12n, 0.06nm)[20].
3) The slow diffusive motion can occur between z=0 (3f and 12n sites) and z=1/2 plane (4h, 12o and 6m sites). The diffusion from 12o site to 12n site occur in LaNi5H5, LaNi5H6 and LaNi5H7, and 12n site to 12o site in LaNi5H6, LaNi5H7 and LaNi5H8, and 3f site to 12o site in LaNi5H5. The only way for a hydrogen atom to escape from the cluster is to jump to a 12o site[21].
In fact, the 12o site seemed to be the “hub” of the process if diffusion occurs by nearest-neighbor jumps, as provided with the experimental study[21]. We can conclude that there must be at least some occupancy of 12o site during absorption. When the amount of hydrogen absorbed varies from 5 to 8, the maximum of hydrogen atoms in 12n site is 3, just as the early experiment results[21].
In La2Ni10H14, the position of hydrogen varies slightly compared with the initial model. The final distribution of hydrogen is 3 at 12d, 2 at 6cⅠ, 1 at 6cⅡ, 1 at 2b, which accords with the earlier experiments[22].
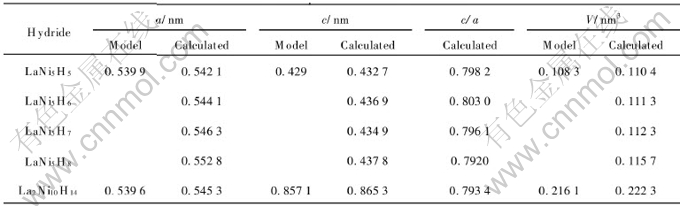
It can also be affirmed from Table 3 that with the increase of the amount of hydrogen, the a and c axis increase and the cell volume expanses. The proportions c/a of hydrides lie in the interval 0.7920-0.8030, quite agreeing with the values ![]() [23].
[23].
3.2 Electronic structure
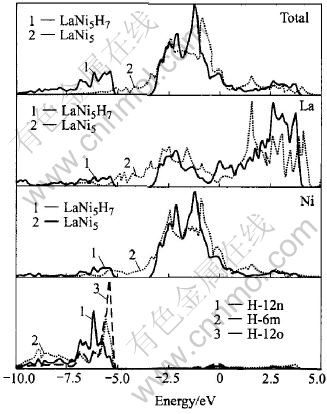
Fig.1 Total and partial densities of states for LaNi5 and LaNi5H7(P6/mmm)
3.2.1 Band structures and density of states
Fig.1 displays the total and partial densities of states (DOS) of LaNi5 and LaNi5H7 with the Fermi level EF as the energy zero. As found in earlier theoretical[24-26] and experimental work[27, 28], the occupied bands of LaNi5 and its hydrides are dominantly but not completely filled by Ni-3d bands. They are also hybridized with lanthanum-contributed (6sp-5d) derived states[27] with EF falling in the rapidly decreasing portion of these bands. There is very little La contribution below EF and the major La DOS component arises above EF centered at 1.57eV in LaNi5 and 2.63eV in LaNi5H7, which is from the 4f states according to Ref.[29]. It can be inferred that the small structure observed in the total DOS both in LaNi5 and its hydride arises from the Ni-La interaction. The energy difference between the Ni-3d electrons and the La-5d electrons is so large that the d electrons are more localized on the Ni-sites[30]. With the addition of hydrogen to LaNi5, a new band appeared, lying at a lower energy than the conduction band of the host metal, which can be filled with the added electrons. During the hydrogenation process electrons are progressively transferred from the conduction band of the host metal into this new band, which centers at -5.37eV. This band involves both the symmetrical (bonding) and the anti-symmetrical (anti-bonding) orbitals derived primarily from the H-1s orbital but with a little d admixture. These states are referred to as hydrogenic states[31]. The hydrogen in LaNi5 hydrides is tending toward the anionic model[32]. The centre of the d-band in hydride shifts downwards from -0.86eV to -1.25eV compared with LaNi5, owing to the entry of the protons which lowers the energy of the d band. Additionally, this band narrows from 2.39eV in LaNi5 to 1.74eV in LaNi5H due to the large lattice expansion upon hydrogenation in part and also to the modification of the Ni-3d bands on account of the Ni-H interaction. The narrowing also explains the shape change of the absorption edge in XANES spectra[7]. A very weak structure at low energies due to the M-H bonding is presented in Figs.1(b) and (c) respectively with a strong Ni-H bonding and a much weaker La-H bond, showing that the hydrogen atoms bind mostly with the nickel atoms[10].
The contour plots (Fig.2) of the charge densities ρ(x) along the c axis in the (21[DD(]-[DD)] 1[DD(]-[DD)]0) plane of both LaNi5 and LaNi5H7 (P6/mmm and P63mc) can also explain it. In Fig.2(a), the high charge density regions are centered at the Ni atoms in contrast to the low charge density at the interstitial regions, which is well delocalized and not significantly directional, indicative of metallic bonding. A relatively high electron-density region extends from the H site, being located within the corresponding low-density regions of the host alloy, towards the Ni site (Figs.2(b) and (c)). On the other hand, there is very weak directional La-H bonding in this figure showing the greater importance of the Ni-H interaction than that of the La-H. No localized charge can be detected, suggesting that the local Ni-H interactions would be more ionic than covalent as shown in Ref.[33].
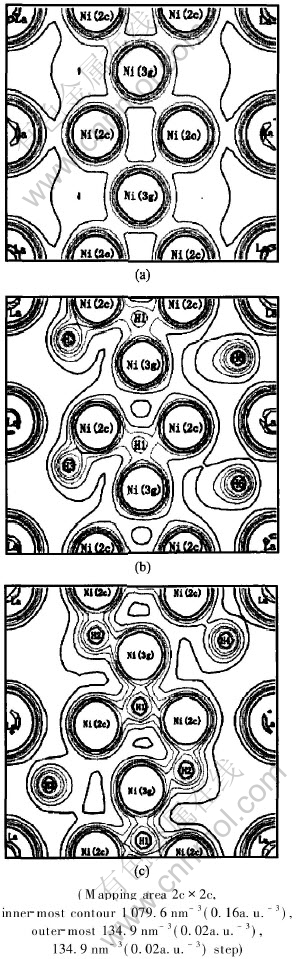
Fig.2 Valence electron density of LaNi5(a),LaNi5H7(P6/mmm)(b)
and La2Ni10H14(P63mc)(c) for (![]() ) plane
) plane
The DOS at Fermi level decreases after hydrogenation (Fig.3) which is probably caused by filling of the new bonding states formed in these compound with the added electrons of hydrogen and part of excited d electron into empty states in s-p band, indicated in Fig.4 that the d-orbital distribution to total DOS at EF reduces with the amount of hydrogen. Whereas the lanthanum d contribution becomes important (Fig.3 and Fig.5) as observed in XANES spectra[9], owing to the Fermi level falling in a rising portion of the DOS of the lanthanum valence electrons, while no contribution in LaNi5[34]. What is more, the contribution of La and H to the DOS increases as the amount of hydrogen increases, in contrast to the decrease of Ni.
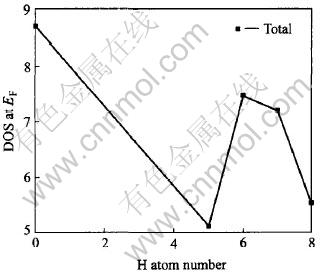
Fig.3 Variation of DOS at Fermi energy level amount of hydrogen added upon hydrogenation
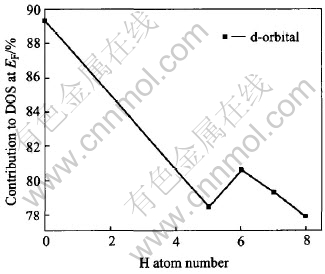
Fig.4 Contribution of d-orbital to total DOS at Fermi level
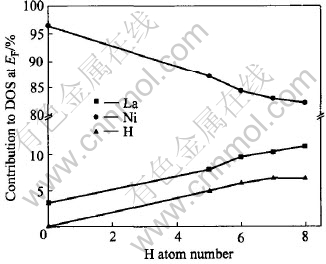
Fig.5 Contribution of different atoms to total DOS at Fermi level
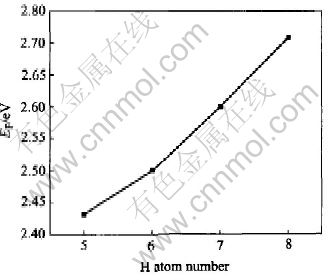
Fig.6 Position of Fermi energy with different amounts of hydrogen into LaNi5
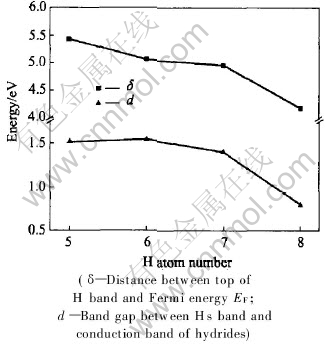
Fig.7 Changes of δ and d values in LaNi5Hx(x=5-8)
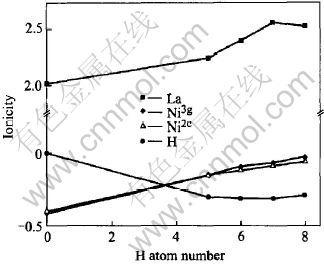
Fig.8 Changes in ionicities of atoms in LaNi5 and LaNi5Hx(x=5-8)
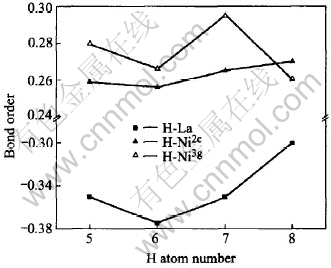
Fig.9 Changes in bond order of H-metal inLaNi5Hx(x=5-8)
The position of Fermi level rises with the addition of hydrogen atoms due to the presence of supplementary electron in part brought by added hydrogen atom and also to the deformation of the nickel d bands by the Ni-H interaction (Fig.6). The δ and d values are plotted in Fig.7, where δ denotes the distance between the top of H band and the Fermi level EF, and d denotes the band gap between Hs band and conduction band of hydrides. The trend of δ and d values is similar as hydrogen capacity increases. Furthermore, there exists a knee point when hydrogen equals 7 and when hydrogen is more than 7 the values changes suddenly. It can be speculated that when hydrogen capacity is larger than 7 the hydrides is less stable.
3.2.2 Mulliken population analysis
The ionicity of each atom in LaNi5 and its hydrides was estimated according to the Mulliken population analysis. As shown in Fig.8, the ionicity of the La is always positive. On the other hand, the ionicities of the H and Ni are always negative[33]. The results reveal significant charge transfer from La to Ni and H. As the amount of hydrogen added increases, the ionicity of H is nearly constant and keeps about 0.3e, whereas, the transferred charge on La increases and the obtained charge on Ni decreases. For example, there is a transfer of about 0.4e per Ni from La to Ni in LaNi5 while only about 0.08e in LaNi5H7 (P6/mmm and P63mc). This slightly shifts the d band of hydrides to lower energies because of the decrease in negative charge, which accords with the calculation of DOS shown in Fig.1.
The bond order is the overlap population of electrons between atoms. This is a measure of the strength of the covalent bond between atoms. The bond order between the hydrogen atom and the La atom is always negative (Fig.9) regardless of hydrogenation, indicating a repulsive interaction operating between them. On the other hand, the bond order on Ni-H is always large and positive. It is clear that the hydrogen atom bonds more strongly with the Ni3g atoms than with Ni2c atom except for x=8 since the ionicity of Ni3g atom is a little higher than Ni2c atom shown in Fig.8. Likewise, the Ni-H bond is stronger than the La-H bond which is consistent with the result of the electron distribution shown in Fig.2. It can be also found a knee point at H capacity equal to 7 and after it the bond order changes severely. Fig.10 shows the change in the bond order between the M-M pairs. The bond order of La-Ni is negative and that of Ni-Ni is positive regardless of hydrogenation. This shows an anti-bonding interaction operating between La and other atoms. Hydrogen atom makes a stronger chemical bond with the Ni atom than with the La atom, despite the larger affinity of La than Ni for hydrogen on the binary metal-hydrogen system. The bond strength between metal pairs is reduced by the introduction of a hydrogen atom into the alloy. Similar results are also reported by Suenobu et al[7]. A platform can be found between x=6 and x=7. Associated with Fig.9, it can be inferred that the influence of hydrogen capacity on hydrides is slight with x lying between 6 and 7 and the hydride is stable.
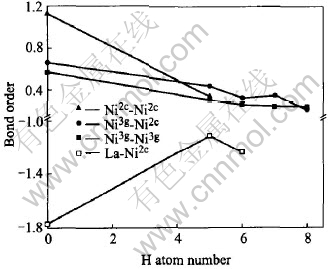
Fig.10 Changes in bond order of metal-metal in LaNi5 and LaNi5Hx(x=5-8)
3.3 Heat of formation
Heat of formation ΔH is crucial to assessing the practical viability of a hydride material and has been a primary focus of experimental work. Since hydrides are not easily amenable to measurement by experiments, theoretical estimates are of interest and value. Pasturel et al[35, 36] have developed semi-empirical equations to estimate the heat of formation of LaNi5-type hydrides starting from the model of Bouten et al[37] and Mal et al[38]. The ΔH value from this model is -29.7kJ/mol. We have calculated the theoretical heat of formation of the hydride directly from the total energies of LaNi5, hydrides and H2. Table 4 lists the total energies E and heat of formation ΔH obtained from our calculations. Treating all the hydrides with the same computational machinery ensures that systematic errors reduce to the greatest extent in energy differences. The theoretical heat of formation per molecular hydrogen ΔH for the hydrogen reaction, yLaNi5+H2=(LaNi5Hx)y+ΔH can be calculated as follows[26, 29]:
![]()
Table 4 Calculated total energies E and heat of formation ΔH
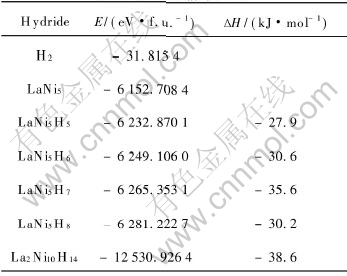
It is apparent that E(H2) is as important to the determination of ΔH as the condensed phase total energies. Including E(Hatom), the total ground state E*(H2) of the H2 molecule from our computation for a (1nm)3 repeated cell is
E*(H2)=E(H2)-2E(Hatom)
=-6.7950eV-2(12.5102eV)
=-31.8154eV
The heat of formation of LaNi5H7 is the lowest in LaNi5Hx(x=5-8) from Table 4, which shows LaNi5H7 is the most stable with the amount of hydrogen equal to 7. This result also accords with the suggestion of DOS and Mulliken population analysis. The value (-35.6kJ/mol) agrees with calorimetric measurements varying from -32.1±0.1kJ/mol at 298K for LaNi5H6.2[39] to -34.8±1.8kJ/mol at 285K for LaNi5H6.33[40]. In contrast with the value from the semi-empirical model[38] and from the theoretical calculation by means of the tight-binding linear muffin-tin orbital method[26], this result is closer to the value from experiments. The method in this work is perhaps more creditable and practicable to obtain theoretical heat of formation of LaNi5-type hydrides. When the amount of hydrogen reaches to 7, the heat of formation of the P63mc structure is lower than that of P6/mmm, which shows the P63mc structure is more reasonable than P6/mmm to β-phase hydrides of LaNi5.
4 CONCLUSIONS
By using the super-cell total energy calculation combined with ultra-soft pseudo-potential the geometry and electronic structure and energetics of LaNi5 and LaNi5Hx (5≤x≤8) were obtained. The hydrogen motion in β-phase hydrides, including long range diffusion and short range hop, can be observed during the process of geometry optimization as found in experiments. A new band was formed during hydrogenation mainly from the bonding between the hydrogen s orbital and the nickel s-p-d orbital. The band gap between this band and the conduction band can be observed clearly. The calculations reveal the charge transfer from La to Ni and H. The hydrogen takes off the localized La-5d electron near the Fermi level and is mainly inclined to bond with Ni. The calculated total energies can deduce the heat of formation for LaNi5Hx (5≤x≤8), which is very favorable agreement with measurements on LaNi5 hydrides. From the analysis of electronic structure and heat of formation, we can conclude that β-phase hydrides of LaNi5 are most stable when hydrogen atom capacity locating in the range of 6 and 7.
Acknowledge
The authors thank Prof. C. M. Hong of Beijing University and Prof. G. Chen of Jilin University for their helpful discussion for this paper.
REFERENCES
[1]Willems J J G, Buschow K H. From permanent magnets to rechargeable hydride electrodes [J]. J Les-Common Met, 1987, 129(1): 13-30.
[2]Bittner H F, Badcook C C. Electrolyte film structure on battery separator and electrode materials [J]. J Electrochem Soc, 1983, 130(2): 259-264.
[3]Willems J J G. Metal hydride electrode electrodes stability of LaNi5-related compounds [J]. Philips J Res, 1984, 34(1): 98.
[4]Deenadas C, Thompson A W, Craig R S, et al. Low temperature heat capacities of laves phase lanthanide-aluminum compounds [J]. J Phys Chem Solids, 1971, 32(8): 1853-1866.
[5]Takeshita T, Gschneidner K A Jr, Thome D K, et al. Low-temperature heat-capacity study of haucke compounds CaNi5, YNi5, LaNi5 and TiNi5 [J]. Phy Rev B, 1980, 21(12): 5636-5641.
[6]Ohlendorf D, Flotow H E. Experimental heat capacities of LaNi5, α-LaNi5H0.36 and β-LaNi5H6.39 from 5 to 300K: Thermodynamic properties of the LaNi5-H2 system [J]. J Chem Phys, 1980, 73(6): 2973-2948.
[7]Sueenobu T, Sakaguchi H, Adachi G. Studies on local structure in hydrogenated amorphous LaNi5 films using extended X-ray absorption fine structure [J]. J Alloys Comp, 1993, 190(2): 273-277.
[8]Suenobu T, Sakaguchi H, Tsuji T, et al. Extended X-ray absorption fine structure studies on local structure in amorphous LaNi5.0 films [J]. Bull Chem Soc Jpn, 1991, 64(12): 3522-3527.
[9]Tryk D A, Bae I T, Hu Y, et al. In situ X-ray absorption fine structure measurements of LaNi5 electrodes in alkaline electrolytes [J]. J Electrochem Soc, 1995, 142(3): 824-828.
[10]Gupta M. Electron properties of LaNi5 and LaNi5H7 [J]. J Less-Common Met, 1987, 130(1): 219-227.
[11]Gupta M. Electronic structure and stability of hydrides of intermetallic compounds [J]. J Alloys and Comp, 1999, 293-295(1): 190-201.
[12]WEI Wen-lou, GUO Jin, DENG Wen, et al. Correlation between electronic structure of LaNi5 base alloys and hydrogen absorption properties [J]. The Chinese Journal of Nonferrous Metals , 2002, 12(3): 501-504.
[13]Percheron-Guégan A, Lartigue C, Achard J C, et al. Neutron and X-ray diffraction profile analyses and structure of LaNi5, LaNi5-xAlx and LaNi5-xMnx intermetallics and their hydrides (Deuterides) [J]. J Less-Common Met, 1980, 74(1): 1-12.
[14]Lartigue C, Percheron-Guégan P, Achard J C. Hydrogen (deuterium) ordering in the β-LaNi5Dx (x>5) phases: a neutron diffraction study [J]. J Less-Common Met, 1985, 113(1): 127-148.
[15]Lartigue C, Le Bail A, Percheron-Guégan A. A new study of the structure of LaNi5D6.7 using a modified rietveld method for the refinement of neutron powder diffraction data [J]. J Less-Common Met, 1987, 129(1): 65-76.
[16]Payne M C, Teter M P, Allan D C, et al. Interative minimization techniques of ab initio total-energy calculations: molecular dynamics and conjugate gradients [J]. Rev Mod Phys, 1992, 64(4): 1045-1097.
[17]Perdew J P, Wang Y. Atoms, molecules, solid and surfaces: applications of the generalized gradient approximation for exchange and correlation [J]. Phys Rev B, 1992, 46(11): 6671-6687.
[18]Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Phys Rev B, 1990, 41(11): 7892-7895.
[19]Kresse G, Furthmüller J. Efficient Iterative schemes for Ab-initio total-energy calculations using a plane-wave basis set [J]. Phys Rev B, 1996, 54(16): 11169-11186.
[20]Lartigue C, Percheron-Guégan A, Achard J C. Study of the different types of hydrogen motion in the β-LaNi4.5Al0.5 hydrides by quasi-elastic neutron scattering [J]. J Less-Common Met, 1984, 101(2): 391-403.
[21]Westlake D G. A geometric model for the stoichiometry and interstitial site occupancy in hydrides (deuterides) of LaNi5, LaNi4Al and LaNi4Mn [J]. J Less-Common Met, 1983, 91(2): 275-292.
[22]Thompson P, Reilly J J, Corliss L M, et al. The crystal structure of LaNi5D7 [J]. J Phys F Met Phys, 1986, 16(6): 675-685.
[23]Gurewitz E, Pinto H, Dariel M P, et al. The structure and dynamics of hydrogen in LaNi5H6 studied belastic and inelastic neutron scattering [J]. J Phys F: Met Phys, 1983, 13(5): 545-554.
[24]Malik S K, Arlinghaus F J, Wallace W E. Calculation of the spin-polarized energy-band structure of LaNi5 and GdNi5 [J]. Phys Rev B, 1982, 25(10): 6488-6491.
[25]Sluiter M, Takahashi M, Kawazoe Y. Theoretical study of phase stability in LaNi5-LaCo5 alloys [J]. J Alloys Comp, 1997, 248(1): 90-97.
[26]Nakamura H, Nguyen-Manh D, Pettifor D G. Electronic structure and energetics of LaNi5, α-La2Ni10H and β-La2Ni10H14 [J]. J Alloys Comp, 1998, 281(1): 81-91.
[27]Weaver J H, Franciosi A, Wallace W E, et al. Electronic structure and surface oxidation of LaNi5, Er6Mn23, and related systems [J]. J Appl Phys, 1980, 51(11): 5847-5850.
[28]Fuggle J C, Hillebrecht F U, Zelelr R, et al. Electronic structure of Ni and Pd alloys(Ⅰ)―X-ray photoelectron spectroscopy of the valence bands [J]. Phys Rev B, 1982, 27(4): 2145-2178.
[29]Hector L G Jr, Herbst J F, Capehart T W. Electronic structure calculations for LaNi5 and LaNi5H7: energetics and elastic properties [J]. J Alloys Comp, 2003, 353(1): 74-85.
[30]Bucur R V, Lupu D. Hydride stability and band structure of the components in the Pd1-xMx and LaNi5-xCux systems [J]. J Less-Common Met, 1983, 90(1): 203-209.
[31]Wallace W E, Pourarian F. Photoemission studies of LaNi5-xCux alloys and relation to hydride formation [J]. J Phys Chem, 1982, 86(6): 4958-4956.
[32]Wallace W E. Bonding of metal hydrides in relation to the characteristic of hydrogen storage materials [J]. J Less-Common Met, 1982, 88(1): 141-157.
[33]Yukawa H, Takahashi Y, Morinaga M. Alloying effects on the electronic structures of LaNi5 containing hydrogen atoms [J]. Intermetallics, 1996, 4(Suppl 1): S215-S224.
[34]Chung Y, Takeshita T, McMasters O D, et al. Influence of the lattice and electronic factors on the hydrogenation properties of the RNi5-base (R is a rare earth) haucke compounds: results of low temperature heat capacity measurements [J]. J Less-Common Met, 1980, 74(1): 217-223.
[35]Pasturel A, Chatillon-Colinet C, Percheron-Guégan A, et al. Thermodynamic and structural properties of LaNi4M compunds and their related hydrides [J]. J Less-Common Met, 1982, 84(1): 73-78.
[36]Pasturel A, Liautaud F, Chatillon-Colinet C, et al. Thermodynamic study of the LaNi5-xCux system [J]. J Less-Common Met, 1984, 96(1): 93-97.
[37]Bouten P C P, Miedema A R. On the heats of formation of the binary hydrides of transition metals [J]. J Less-Common Met, 1980, 71(1): 147-160.
[38]Van Mal H H, Buschow K H J, Miedema A R. Hydrogen absorption in LaNi5 and related compounds: experimental observations and their explanation [J]. J Less-Common Met, 1974, 35(1): 147-160.
[39]Hubbard W N, Rawlins P L, Connick P A, et al. The standard enthalpy of formation of LaNi5 and the enthapies of hydriding of LaNi5-xAlx [J]. J Chem Thermodynam, 1983, 15(4): 785-788.
[40]Murray J J, Post M L, Taylor J B. The thermodynamics of the LaNi5-H2 system by differential heat flow calorimetry (Ⅱ): The α and β single-phase regions [J]. J Less-Common Met, 1981, 80(1): 211-219.
Foundation item: Project(2001CCA05000) supported by the National Key Program for Basic Research; Project(2002CB211800) supported by the National Basic Research and Development Program of China
Received date: 2004-10-20; Accepted date:2005-04-04
Correspondence: WU Feng, Professor, PhD; Tel: +86-10-68912508; Fax: +86-10-68451429; E-mail: wufeng863@vip.sina.com

