���±�ţ�1004-0609(2011)08-2017-06
TiO2�ɼ����H2O2����ˮ�е��л���Ⱦ��
�ƽ���1����С��2���� ԭ1�������2���ܿ���2
(1. ����ְҵ����ѧԺ �����й�ҵ��ˮ�������ˮ��Դ�������ص�ʵ���ң����� 518055��
2. ���ϴ�ѧ ұ���ѧ�빤��ѧԺ����ɳ 410083)
ժ Ҫ���Ի�����X-3B��������ģ����Ⱦ��о���������(���ѿ�ͽ��ʯ)��H2O2��TiO2�ɼ�������ܵ�Ӱ�죬���о�Fe3+����һ��Ӧ��ϵ�е�ЭͬЧӦ�������������H2O2���������£����ѿ�ͽ��ʯTiO2���ܿɼ�������������X-3B����TOC��ȥ���ʴ�65%����Ӧ�������ڷ�ɫ���ŵ��ƻ�������TiO2��������λ����CO32-�ﵽ���ͺ���Ի�����X-3B��������Ч����TiO2�ܿɼ����H2O2���ⱽ�ӣ����ʯTiO2��ʾ�������ѿ�TiO2���ߵĴ����ԣ���Ӧ120 min�Ա��ӵĽ����ʴ�80%���ҷ�Ӧ��ϵ���ɵĻ�״�м�����Ũ�����Խ����ѿ�TiO2�ĸߣ���Fe3+ЭͬTiO2�ɼ����H2O2���ⱽ��ʱ����ӦЧ�������ӿ죬��20 min��Ӧ���Ա��ӵĽ����ʿɴ�100%�������ʯTiO2��ʾ��Ϊ���Ե�ЭͬЧӦ��
�ؼ��ʣ��ɼ�������������ѣ��������⣻ЭͬЧӦ��������X-3B������
��ͼ����ţ�O 643.32���� ���ױ�־�룺A
Degradation of organic pollutants with H2O2 photocatalyzed by TiO2 under visible irradiation
TANG Jian-jun1, FAN Xiao-jiang2, ZOU Yuan1, LIU Shu-jun2, ZHOU Kang-gen2
(1. Shenzhen Key Laboratory of Industrial Water Conservation and Municipal Wastewater Resources Technology,
Shenzhen Polytechnic, Shenzhen 518055, China;
2. School of Metallurgical Science and Engineering, Central South University, Changsha 410083, China)
Abstract: The effects of crystalline structure (anatase and rutile) and H2O2 on TiO2 visible-light photocatalysis were studied, and the synergistic effect of Fe3+ in the reaction system was also studied using the reactive red X-3B and phenol as model pollutants. The results indicate that reactive brilliant red X-3B can be photocatalytic degradated by anatase or rutile TiO2 under visible-irradiation with H2O2 addition, the TOC removal rate can reach 65%, and the degradation process is not limited to the broke down of chromophore groups, however, when the surface sites of TiO2 photocatalyst (anatase or rutile) is preeminently occupied by CO32-, the photodecomposition of reactive brilliant red X-3B can not be processed. The phenol can be degradated with H2O2 catalyzed by TiO2 visible-light photocatalysis, the rutile form exhibit a better photocatalytic activity, its degradation rate can reach 80% after 120 min reaction, and the concentration of the ring intermediates detected during phenol decomposition is evidently higher when using rutile TiO2 as photocatalyst. The snergistic degradation of phenol with H2O2 photo-catalyzed by TiO2 and Fe3+ is evidently fast, the degradation rate of phenol can reach 100% after 20 min reaction, and rutile TiO2 show more evident synergistic effect.
Key words: visible light photocatalysis; titanium dioxide; hydrogen peroxide; synergistic effect; reactive brilliant red X-3B; phenol
TiO2�����Ϊһ�ֻ����ǻ����ɻ�(��OH)���̵ĸ�������������Ӧ���ڽ���ˮ�е��л���Ⱦ�﷽���ܵ����ǵĹ�ע����Ŀǰ������һЩؽ������IJ���[1]��һ��TiO2�Ĺ���Ӧ��Χ�����ڲ���С��387 nm������������̫�������д˲�����������5%������TiO2������⼤���²����ĵ��ӿ�Ѩ��(e-h+)���������¸��ϣ����Ч�����ޡ�Ϊ�ؿ�TiO2�Ĺ���Ӧ��Χ���ɼ�������һ�Ƕ�TiO2����[2]��ǽ���[3]���ӣ������Ӵ�������ؿ�TiO2�Ĺ����շ�Χ���ɼ�������Ҳ��ͬʱӰ��TiO2���ȶ��ԣ����ǻ��ڹ������ķ�Ӧ������չTiO2�ɼ������Ӧ[4]��������һ��Ӧ�У���������(��Ⱦ����Ⱦ�ﱾ��)�ķ�ɫ���ű��ƻ���Ӧ��Ӧֹͣ����ˣ�����Ⱦ��Ľ��ⲻ���ס�Ϊ���TiO2�����Ч�ʣ���ʵ���о���Ӧ���У���������Ӧ��ϵ����H2O2[5]��H2O2��һ�������ĵ��ӷ��������������TiO2���⼤���������e-h+�Եĸ��ϣ��Ӷ���øߴ�Ч�ʣ��������о�һ��ֻ��������������
H2O2��������TiO2�����γɸ����������ؿ�TiO2�Ĺ����շ�Χ���ɼ�����[6-7]��TiO2�ܿɼ����H2O2����ˮ����[6]��[8-9]�ȷ�Ⱦ�����л���Ⱦ��ƽ�����[8-9]�о���������H2O2���������£��ɼ�������ⱽ��ʱ�����ʯTiO2��ʾ�����ѿ�TiO2���쾧TiO2(P25 TiO2)���и��ߵĴ����ԡ����о�ָ��[10-11]�����ִ����ԵIJ��������ڷ�Ӧ��ϵ���ɲ�ͬ���͵Ļ������֣�������漰��ͬ�ķ�Ӧ;�����¡�
��TiO2��H2O2���������µĿɼ������Ӧ��ȥ��ˮ����е��л���Ⱦ�������������ƣ��Ȳ���Ҫ���ӵIJ��Ӵ�����Ӱ���������ȶ��ԣ��ҷ�Ⱦ�����л���Ҳ���ڴ������µõ����⡣Ȼ����Ŀǰ�����������һ��Ӧ���о����������������������о�Ҳͬʱ����[8-9]����һ��Ӧ�����ڷ�Ӧ���ʽϵͼ�H2O2�����ʵ͵Ȳ��㡣
Ϊ�ˣ����о������ѿ��ʯTiO2����������Ի�����X-3B��������ģ����Ⱦ�ͨ���Բ�ͬ��Ӧ�����µĶԱ��о���������ͨ������Ӧ��ϵ����Fe3+���γ�ЭͬЧӦ�����ڼ������һ��Ӧ���˽⣬�������DZ��Ӧ��ʱ�IJ��㣬Ϊ����ˮ�����е�ʵ��Ӧ���ṩ���������뼼��֧�֡�
1 ʵ��
1.1 ��������Ʊ�
ʵ�����õ�TiO2��������������Ķ���Ϊǰ���ˮ����������Ʊ�������μ�����[12]���������õ����ѿ���TiO2�������¶�Ϊ400 �桢����ʱ��Ϊ2 h�����ΪTIO-A�������ʯ��TiO2�������¶�Ϊ900 �桢����ʱ��Ϊ2 h�����ΪTIO-R��
��������CO32-��TiO2���Ʊ�������Ϊ����5.0 g��TIO-A��TIO-R�ֱ��������50 mL��Ũ��Ϊ 1 mol/L��Na2CO3��Һ�У���̬�½���24 h����0.45 ��m������Ĥ���˳�������壬�������¶�70 ��ĸ������к�24 h����ϸ���ã��ֱ����TIO-A-CO3��TIO-R-CO3��
1.2 ���ʵ��
������Դ����CMH-250��±�ƹ�������װ��(��ʦ���ѧ������)������200 W����Դ������Χ380-800 nm���˹�Ƭ���Ϊ400 nm����ǿΪ75.9 W/m2��ʵ���������ΪVis������XRD��BET���Խ����ʵ�����õĹ����TIO-A��TIO-R�������������1���С�ģ����Ⱦ��ѡ�û�����X-3B(����Sigma��˾��Ʒ)������(AR)����ʼŨ�Ⱦ�Ϊ15 mg/L����Һ���50 mL����Ũ��1 mol/L��NaOH��1:25 ��HNO3��Һ����pH��3������Ĵ���TiO2(TIO-A��TIO-R)��Ũ��Ϊ1.0 g/L��Fe3+(FeCl3��AR)��Ũ��Ϊ34 mg/L��H2O2��ʼŨ��Ϊ1.1 mmol/L��
��1 TiO2���������������
Table 1 Characteristics parameters of TiO2 photocatalysts

ʵ������У����Ƚ���ģ����Ⱦ�P����(TIO-R��TIO-A��Fe3+)������Һ�ڰ�̬�·�ɢ30 min�������ڷ�Ӧ���н��й��ʵ�飻��Ӧһ��ʱ���ȡ����ˮ���Ⱦ�Hitachi CR22G������䶳���Ļ����룬����0.45 ?m������Ĥ���˺�����Ũ�ȷ�����
1.3 ��������
XRD��������X��Pent Pro��X���������ǣ����£�Cu K��Դ��40 kV��40 mA��X���߲�����=1.540 6 nm������Scherrer��ʽ������Ʒƽ��������BET�ȱ������������3H-2000ȫ�Զ��������ȱ����ǡ�
������X-3BŨ�ȷ�������Hitachi U-3010�� ��-�ɼ��ֹ��ȼ�(��max=536 nm)�����Ӽ��併������м�����Ũ�ȷ�������Waters1525��ЧҺ��ɫ���ǣ����н�����10 ?L��������V(�״�):V(ˮ)=25:75������0.8 mL/min��������SYMMETRY C18��4.6 mm��150mm��2487��������˫ͨ���������������Ӻ��ڱ����ӵļ�Ⲩ��Ϊ270 nm��ͣ��ʱ��ֱ�Ϊ15.267��7.258 min���������Ա����ӵļ�Ⲩ���ֱ�Ϊ246��289 nm��ͣ��ʱ��Ϊ5.139��3.319 min�����л�̼(TOC)��������Shimadzu TOC-VCPH��TOC�����ǣ���������TC��������Ӧ�¶�680 �档
2 ��������
2.1 ������X-3B�Ľ���ʵ��
���ǵ�H2O2��������X-3B��CO32-��TiO2�����������λ����ͬ�ģ���CO32-���������ܸ�ǿ��Ϊ���Ʊ���������CO32-�Ĺ����TIO-A-CO3��TIO-R-CO3��ʵ�鷢�֣�TIO-A��TIO-R�ڱ�������CO32-��H2O2�ͻ�����X-3B����������Ч����
ͼ1��ʾΪ������X-3B�ڲ�ͬ��Ӧ�����µĿɼ�����������������H2O2�ڲ�������400 nm�Ŀɼ��⼤���£��Ի�����X-3B�������ã���ԭ����H2O2ֻ�����ղ���С��320 nm�������߶��ֽ������OH[13]����Vis/O2��Vis/H2O2�����£���TIO-A�������ʱ���ܺܺý��������X-3B����Ӧ75 min�����ʼ���100%������TIO-A-CO3�������ʱ���������TIO-A��������λ��CO32-���ͺ���Ի�����X-3B�����������ã�֤ʵȾ�����л�����ɼ��������ı�Ҫ���������������ڹ�����ı���[14]����Vis/O2�����£�TIO-R�Ĵ�����������Ӧ75 min�Ի�����X-3B�Ľ����ʻ�����15%��������Ӧ��ϵ����H2O2ʱ��TIO-R��90 min�Ի�����X-3B�Ľ����ʽ�100%���������TIO-R��������λ��CO32-���ͺ���TIO-R-CO3�������ʱ���Ի�����X-3BҲ��������Ч����TiO2�ɼ��������Ⱦ�����л�����һ��Ӧ��ʵ����H2O2�����ڹ����TiO2�����γɸ����������ؿ�TiO2�Ĺ����շ�Χ���ɼ������� ��[8]����H2O2���������£�TIO-A-CO3��TIO-R-CO3�Ի�����X-3B���������ã���ԭ����TIO-A��TIO-R�����������λ��CO32-ռ�ݺ�H2O2����������TiO2�����γɸ����

ͼ1 ��ͬʵ�������»�����X-3B�Ŀɼ�����������
Fig.1 Photodegradation of reactive red X-3B under visible light irradiations: (a) TIO-A; (b) TIO-R
ͼ2��ʾΪ��ͬ��Ӧ�����»�����X-3B�����UV-Vis�����ס���ͼ2�ɿ�������Vis/O2��Ӧ��������TIO-A�������ʱ����������X-3B�Ľ����ʴ�100%ʱ����UV-Vis�������ڦ�=538 nm�����������շ���ʧ����������X-3B�з�ɫ��ż��������������������������Ȼ���(200~350 nm)�����շ��������������ڽӽ�200 nmʱ�����к�ǿ��ĩ�����գ���Vis/H2O2��Ӧ�����£���TIO-A��TIO-R�����������������X-3B�Ľ����ʴ�100%ʱ��������=538 nm�����������շ���ʧ���Ҵ��������������Ȼ��ŵ����շ�Ҳ�и���̶ȵļ�������ͼ3��ʾ����Vis/H2O2�����½��������X-3Bʱ��TIO-A��TIO-R��TOC��ȥ���ʷֱ�ԼΪ65%��62%������Vis/O2��Ӧ������ʱ����TIO-A�Ի�����X-3B�Ľ����ʴ�100%����TOC��ȥ���ʽ�30%��

ͼ2 ��ͬ��Ӧ�����½��������X-3B��UV-Vis���չ���
Fig.2 UV-Vis absorption spectra of reactive red X-3B photocatalytic degradation at different reaction conditions

ͼ3 ��ͬ��Ӧ�����´����������X-3B��TOCȥ����
Fig.3 TOC removal rate of reactive red X-3B photocatalytic degradation at different reaction conditions
��Vis/O2��Ӧ�����£��ɼ�������������X-3Bʱ����ɫ�IJ�����������ȱ��ƻ����������������ȵĿ�����Ӧ�����Խ��У������ڻ�����X-3B�Ľ�����ͨ�����������ƽ��еģ�����ɫ���ű���ȫ�ƻ���Ӧ��Ӧֹͣ����˻�����X-3B�Ľ��ⲻ���ף�TOCȥ���ʵ͡���Vis/H2O2��Ӧ�����£����������X-3Bʱ������H2O2��������TiO2�����γɸ����������ؿ�TiO2�Ĺ����շ�Χ���ɼ���������˼�ʹ��ɫ���ű���ȫ�ƻ��������ⷴӦ���ɼ������У�������X-3B�Ľ����Ϊ���ף�TOC��ȥ����Ҳ�����ߡ�
2.2 ���ӵĽ���ʵ��
ͼ4��ʾΪTIO-A��TIO-R�ɼ����H2O2���ⱽ���Լ�����������ɵĻ�״�м�����Ũ�ȱ仯����ͼ4�ɿ�������Vis/H2O2�����½��ⱽ��ʱ��TIO-R�Ĵ��������Ժ���TIO-A�ģ���Ӧ120 min��TIO-A��TIO-R�Ա��ӵĽ�����ԼΪ48%��80%���Ը�ЧҺ��ɫ���ܲ��Ե��ڱ��ӵĽ�����������ɶ��ֻ�״�м�����Ա�����(hydroquinone HQ)������(benzoquinone BQ)���ڱ�����(Catechol)�ȣ������ǵ�Ũ�ȱ仯���ƾ�Ϊ���������潵�ⷴӦ�Ľ��ж���С������TIO-A�������ʱ�����Ե��Ļ�״�м����Ũ�Ƚϵͣ������Ũ�ȶ���0.5 mg/L���£�����TIO-R�������ʱ������Ե��Ļ�״�м����Ũ�Ƚϸߣ����ڱ����ӡ��Ա����Ӻͱ��������Ũ�ȷֱ��1.95��1.0��0.6 mg/L��

ͼ4 ��ͬ����TiO2��400 nm H2O2�����¹�����ⱽ�Ӽ���״�м�����Ũ�ȱ仯
Fig.4 Phenol decomposition and formation of ring products of phenol decomposition under visible light irradiations (�ˣ�400 nm): (a) Anatase TiO2; (b) Rutile TiO2 photocatalysts
��TIO-AΪ�����ʱ����Ӧ��ϵ��H2O2������TIO-A�����γ�on-top��?-peroxide�ṹ�����ɼ��⼤�������ɵĻ��������DZ��桤OH���ɻ����������ɻ�O2��-[10]�����ڱ��桤OH���ɻ���������TIO-A�ı��棬���ӵĽ��ⷴӦ��Ҫ�����ڹ�����ı��棬����������������TIO-A���沢���й����Ӧֱ���������С���ӣ��ʷ�ӦЧ�ʵͣ���Ӧ��ϵ�ܲ��Ե��Ļ�״�м����Ũ�ȵ͡�����TIO-R�������ʱ��H2O2������TIO-R��������Ҫ�γɦ�2-peroxide�ṹ�����ɼ��⼤�������ɵĻ������������롤OH���� ��[10]����ˣ����ӵĽ��ⷴӦ��Ҫ��������Һ���ӦЧ�ʸߣ���Ӧ��ϵ�ܲ��Ե��Ļ�״�м����Ũ�Ƚϸߡ�
ͼ5��ʾΪ�����ڲ�ͬʵ�������µĿɼ���������������ͼ5���Կ�������û��H2O2���������£�����TiO2�����(TIO-A��TIO-R)��Fe3+�Ա��ӻ����������ã�����Ӧ��ϵ����Ũ�ȴ��ڲ���5%�ļ��٣�����ΪTiO2�������Ի�Fe3+��������������ģ�TiO2(TIO-A��TIO-R)���ܿɼ����H2O2���ⱽ�ӣ��������Խϵͣ�����ͬ��Ӧ�����£�Ũ��Ϊ1 g/L��TIO-A��TIO-R��120 min��Ӧ�Ա��ӵĽ�����ԼΪ48%��80%����Ũ��Ϊ34 mg/L��Fe3+��80 min��Ӧ�Ա��ӵĽ����ʴ�100%����Fe3+ЭͬTiO2 (TIO-A��TIO-R)�ɼ����H2O2���ⱽ��ʱ����Ӧ���������ӿ죬���Դ������ߵ�������ʱ�ļӺͣ���20 min��Ӧ������ȫ���ⱽ�ӣ���TIO-R��ʾ����TIO-A��Ϊ���Ե�ЭͬЧӦ��
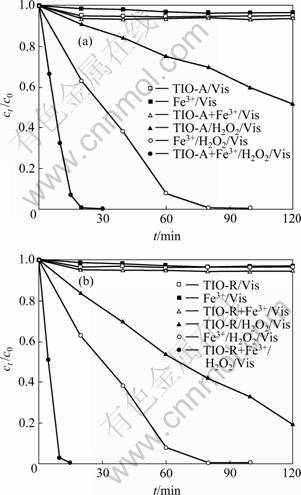
ͼ5 ��ͬʵ�������±��ӵĿɼ�����������
Fig.5 Photodegradation of phenol under visible light irradiation: (a) TIO-A; (b) TIO-R
Fe(OH)22+ Fe2++ ��OH (1)
Fe2++ ��OH (1)
dye dye*
dye*
dye*+Fe3+��dye+ + Fe2+ (2)
����Fenton��Ӧ��ʵ�ʾ��ǽ���Fe3+/Fe2+ѭ ��[15]����Fe(OH)22+����ϵ������Ĺ����շ�Χ�����ڲ�������400 nm��������[16]���ڱ��о��Ŀɼ�������ⱽ�ӵķ�Ӧ�У�Fe2+������ͨ����������;��������������������Ӧ��ϵ��������ЭͬЧӦ��ԭ���ǣ�H2O2������TiO2(TIO-A��TIO-R)�����γɸ�������ɼ��⼤��������������Ӧ����һЩ��ԭ���м���(�����Ӽ�������)����ԭ���м�����Fe3+�γɸ������Fe3+�Ļ�ԭ���ó�ΪFe2+��������Ҫ; ��[17-18]���Ӷ��ٽ�Fe3+/Fe2+ѭ�����γɶ������ķ�Ӧ;�����ﵽ��߷�Ӧ���ʼ�H2O2�����ʵ�Ŀ�ġ� TIO-R��ʾ����TIO-A��Ϊ���Ե�ЭͬЧӦ����ԭ��������ΪTIO-R��TIO-A�ɼ����H2O2���ⱽ�ӵķ�Ӧ�漰��ͬ�ķ�Ӧ;����ǰ����Ҫ��������Һ���Ӧ��ϵ���ɵ��м����Ũ�ȸ���(��ͼ4)��
3 ����
1) ��TiO2�ɼ�������������X-3B�У���Ӧ��ʵ�����м����H2O2�����ڹ����TiO2�����γɸ����������ؿ�TiO2�Ĺ����շ�Χ���ɼ��������£�����Ӧ��ϵ����H2O2ʱ�����ʯ TiO2Ҳ��ʾ�ߴ����ԣ���Ӧ90 min�Ի�����X-3B�Ľ����ʴ�100%���ҷ�Ӧ���̲������ڷ�ɫ���ŵ��ƻ���TOC��ȥ���ʴ�65%��
2) �������TiO2(���ѿ����ʯ)��������λ����CO32-�ﵽ���ͺ���Ի�����X-3B�����������ã��ڻ�����X-3B�Ŀɼ������������У������TiO2�Ի�����X-3B��H2O2�����������Ƿ�Ӧ������ǰ�ᡣ
3) TiO2(TIO-A��TIO-R)�ܿɼ����H2O2���ⱽ�ӣ���TIO-R�������ʱ����ʾ��TIO-A���ߵĴ����ԣ��ҷ�Ӧ��ϵ���ɵ��м����(�����Ӽ�������)Ũ�ȸߡ�
4) ��Fe3+ЭͬTiO2(TIO-A��TIO-R)�ɼ����H2O2���ⱽ��ʱ����Ӧ���������ӿ죬���Դ������ߵ�������ʱ�ļӺͣ���TIO-R��ʾ����TIO-A��Ϊ���Ե�ЭͬЧӦ��
REFERENCES
[1] ����ΰ, �����, ��Ӧƽ, �Խ���, ����ƽ. �ɼ����յ�TiO2������о���չ[J]. ��ѧͨ��, 2003, 48(21): 2199-2204.
JI Hong-wei, MA Wan-hong, HUANG Ying-ping, ZHAO Jin-cai, WANG Zheng-ping. Researches in TiO2 photocatalytic induced by visible-light[J]. Chinese Science Bulletin, 2003, 48(21): 2199-2204.
[2] OHNO T, TAGAWA F. Photocatalytic oxidation of water by visible light using ruthenium-doped titanium dioxide powder[J]. J Photochem Photobioa A: Chem, 1999, 127: 107-110.
[3] IHARA T M, IRIYAMA M Y. Visible- light-active titanium oxide photocatalyst realized by an oxygen-deficient structure and by nitrogen doping[J]. Appl Catal B: Environ, 2003, 42: 403-409.
[4] WU T X, LIN T, ZHAO J C, HIDAKA H, SERPONE N. TiO2-assisted photodegradation of dyes. 9. photooxidation of a squarylium cyanine dye in aqueous dispersions under visible light Irradiation[J]. Environ Sci Technol, 1999, 33: 1379-1387.
[5] ������, ������, ������, �� ��, �Խ���, ������. TiO2�������ˮ���ڷ��ڸ�����17��-�ƶ���[J]. ������ѧ, 2007, 28(1): 120-125.
LI Qing-song, GAO Nai-yun, MA Xiao-yan, WANG Li, ZHAO Jian-fu, LIN Le-sheng. Photocatalytic fndocrine disruptor 17��-Estradiol(E2) in drinking water by nano titanium suspended system[J]. Enviromental Science, 2007, 28(1): 120-125.
[6] LI X Z, CHEN C C, ZHAO J C. Mechanism of photodecomposition of H2O2 on TiO2 surfaces under visible light irradiation[J]. Langmuir, 2001, 17: 4118-4122.
[7] OHNO T, MASAKI Y, HIRAYAMA S. TiO2 photocatalyzed epoxidation of 1-decene by H2O2 under visible light[J]. J Catal, 2001, 204: 163-168.
[8] �ƽ���, ��С��, �� ԭ, �˰���, �� ΰ, �ܿ���. H2O2��TiO2�ɼ������Ӧ�е����û���[J]. �й���ɫ����ѧ��, 2009, 19(2): 292-297.
TANG Jian-jun, FAN Xiao-jiang, ZOU Yuan, DENG Ai-hua, ZHANG Wei, ZHOU Kang-gen. Effects of H2O2 on TiO2 photocatalysis under visible light irradiation[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(2): 292-297.
[9] �ƽ���, ��С��, �� ԭ, �˰���, �� ΰ, �ܿ���. TiO2�ľ��Ͷ���ɼ�������ܵ�Ӱ��[J]. �����Ƽ���ѧѧ��, 2009, 31(4): 418-422.
TANG Jian-jun, FAN Xiao-jiang, ZOU Yuan, DENG Ai-hua, ZHANG Wei, ZHOU Kang-gen. Effect of crystall ine structure on TiO2 photocatalysis under visible light irradiation[J]. Journal of University of Science and Technology Beijing, 2009, 31(4): 418-422.
[10] HIRAKAWA T, YAWATA K, NOSAKA Y. Photocatalytic reactivity for O2���� and OH�� radical formation in anatase and rutile TiO2 suspension as the effect of H2O2 addition[J]. Appl Catal A: General, 2007, 325(1): 105-111.
[11] �ƽ���, ��С��, �� ԭ, �� ΰ, �ܿ���. ���ɻ����Ƽ��Բ�ͬ����TiO2�����Ӧ��Ӱ��[J]. ����������ѧѧ��, 2009, 36(3): 36-40.
TANG Jian-jun, FAN Xiao-jiang, ZOU Yuan, ZHANG Wei, ZHOU Kang-gen. Effects of hydroxyl free radical inhibitors on TiO2 photocatalysis with different crystalline structure[J]. Journal of Beijing University of Chemical Technology, 2009, 36(3): 36-40.
[12] �ƽ���, ������, л�ƽ, �� ԭ, �ܿ���. N/TiO2��������Ʊ������ӹ��̷���[J]. ���̹���ѧ��, 2008, 8(1): 172-176.
TANG Jian-jun, WANG Yue-jun, XIE Wei-ping, ZOU Yuan, ZHOU Kang-gen. Preparation of N-doped TiO2 photocatalyst and analysis of doping process[J]. The Chinese Journal of Process Engineering, 2008, 8(1): 172-176.
[13] HERRERA F, KIWI J, LOPEZ A, NADTOCHENKO V. Photochemical decoloration of remazol brilliant blue and Uniblue A in the presence of Fe and H2O2[J]. Environ Sci Techno1, 1999, 33: 3145-3151.
[14] �� ٩, ��Ӣ��, Ҷ��ϼ. ��ͬ��Դ��TiO2�������Ⱦ����Ⱦ���Ӱ��[J]. ̫����ѧ��, 2005, 26(1): 39-43.
WANG Kan, CHEN Ying-xu, YE Fen-xia. Photodegradation of dye pollutants over TiO2 particles under UV-Vis light irradiation[J]. Acta Energiae Solaris Sinica, 2005, 26(1): 39-43.
[15] WALLING C, GOOSEN A. Mechanism of the ferric ion catalytised decomposition of hydrogen peroxide: Effects of organic substrates[J]. J Am Chem Soc, 1973, 95: 2987-2991.
[16] XIE Y D, CHEN F, ZHAO J C. Photoassisted degradation of dyes in the presence of Fe3+ and H2O2 under visible irradiation[J]. J Photochemistry and Photobiology A: Chemistry, 2000, 136: 235-240.
[17] DU Y X, ZHOU M H, LEI L C. Role of the intermediates in the degradation of phenolic compounds by Fenton-like process[J]. J Hazard Maters, 2006, 136: 859-863.
[18] ������, ��Ծ��, ������, ���ֳ�. ��Fenton������ȱ�����Fe�Ļ�ԭ������;����������[J]. �㽭��ѧѧ��: ��ѧ��, 2005, 39(10): 1618-1622.
DU Ying-xun, NIAN Yue-gang, ZHOU Ming-hua, LEI Le-cheng. Reduction of Fe3+ during 4-chlorophenol degradation by Fenton-like process: Role of two reduction pathway[J]. Journal of Zhejiang University: Natural Science, 2005, 39(10): 1618-1622.
(�༭ ����)
������Ŀ���㶫ʡ��Ȼ��ѧ����������Ŀ(2409K3080018)�������пƼ��ƻ�������Ŀ(07K164D0)
�ո����ڣ�2010-06-10�������ڣ�2010-11-20
ͨ�����ߣ��ƽ����������ڣ���ʿ���绰��0755-26731157��E-mail: tangjj7384@oa.szpt.net

