J. Cent. South Univ. Technol. (2007)02-0186-05
DOI: 10.1007/s11771-007-0037-9 
First-principle investigation on stability of Co-doped spinel ��-Mn4-xCoxO8
HUANG Ke-long(�ƿ���)1, CHEN Chun-an(�´���)1, LIU Su-qin(������)1,
LUO Qiong(�� ��)1, LIU Zhi-guo(��־��)2
(1. College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China;
2. Department of Chemistry and Chemical Engineering, Hunan University of Technology, Zhuzhou 412007, China)
Abstract: The mechanism of stability of Co-doped spinel ��-MnO2 that is referred to as spinel LixMn2O4 (x=0) was studied by using the first-principle calculation method. The total energy and formation enthalpy can be decreased remarkably due to the Co substation, resulting in a more stable structure of ��-MnxCr2-xO4. The bond order and DOS analysis were given in detail to explain the nature of stability improvement. The calculated results show that as the content of Co dopant increases, the bond order of Mn��O becomes larger and the peak of density of states around Fermi level shifts toward lower energy. The charge density distribution illustrates that the Mn��O bonding is ionic and partially covalent, and the covalent Mn��O bonding becomes stronger with the increase of Co dopant content. The results confirm that the Co-doping will enhance the stability of ��-MnO2 and hence improve the electrochemistry performance of LixMn2O4.
Key words: first-principles; stability; electrochemical performance; Co-doped ��-MnO2
1 Introduction
Despite the fact that spinel LiMn2O4 possesses a series of attractive characteristics such as high cell voltage, large durability under extreme temperature circumstance and low cost, there are still some open questions arising from its practical application in cathode of lithium ion battery[1-3]. Some studies were reported that the structure of cathode LixMn2O4 becomes less stable when lithium is almost completely removed during charging and host material transforms to ��-MnO2[1,4]. These changes result in the decrease of charging- recharging circles. Therefore the formation of ��-MnO2 during the charging of spinel LiMn2O4 cathode has a side effect on electrochemistry performance of LiMn2O4.
In order to improve the performance of LiMn2O4 cathode, a great deal of experimental work has been conducted by doping a small amount of Co, Cr or Ni to substitute Mn[5-8]. All kinds of models are constructed to account for the mechanism of the transition metal-doped spinel LiMn2O4 based on the first-principles, but most are focused on the correlation between the total energy and the intercalation voltage[9-10]. Although some of them can give insight into understanding the electronic attributes of spinel LiMn2O4, a little information about the stability of the metal-doped LixMn2O4 is acquired especially at the time when x nearly reaches zero. In this study, the effect of Co on the stability of ��-MnO2 was explored.
2 Computational methods
2.1 Method details
The CASTEP package, an implementation of the first principle plane-wave pseudopotential, was designed for simulating the material properties[11]. The DFT that is highly successful for many classes of systems forms the basis for the CASTEP package[12]. The GGA-PW91 functional is specified to describe the system because it generally produces a more accurate and reasonable result after different functionals, including LDA, GGA and meta-GGA, and was carefully evaluated in the system[10].
The electronic wavefunctions, determined by 340 eV cut-off, were expanded to the extent that is sufficient for leading to the convergence with respect to the total energy and forces acting on the atom. The energy reaches the convergence with the accuracy of 2��10-6 eV/atom and meanwhile the force is converged with the accuracy of 0.025 eV/nm. The Monkhorst-Pack k-points 5��5��6 were assigned to define the accuracy of the sampling[13]. No observed Jahn-Teller effect is found in the doped ��-MnO2 as Mn is expected to appear with formal valence of +4. Consequently, the Jahn-Teller effect is not taken into account in this case[13-14].
2.2 Geometric structure and calculation models
The spinel ��-MnO2 belongs to space group Fd-3m.
It is experimentally observed that in spinel ��-MnO2, Mn occupies Wyckoff site of 16d and 32e for oxygen atoms[15]. The different sites are clearly identified by the label of OI, OII, OIII, O�� and M1, M2, M3, M5, M6, M7, M8(Fig.1).
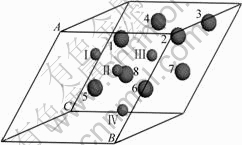
Fig.1 Primitive cell of M4O8 (M represents metal atom (Co or Mn))
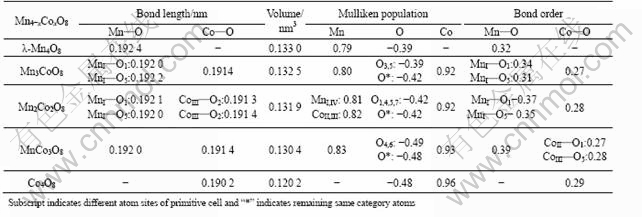
A primitive cell of ��-MnO2 containing four Mn atoms and eight O atoms was employed in the study, and then Mn atoms in cell were substituted by Co atoms to model stoichiometric Mn4-xCoxO8 (x=0, 1, 2, 3, 4). During the calculations, the unit cell of Mn4-xCoxO8 was maintained as the cubic symmetry of host. The structural parameters of Mn4-xCoxO8 are optimized by full relaxation of the cell and the calculated results are presented in Table 1.
Table 1 Characteristic property of various Co-dopant primitive M4O8 cells
3 Results and discussion
3.1 Total energy and formation enthalpy
The total energy is calculated to obtain the formation enthalpy. The formation enthalpy is given by the difference in total energy between MnxCo4-xO8 and the sum of oxides Mn4O8 and Co4O8, i.e.
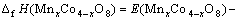
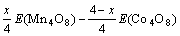 (1)
(1)
where E(MnxCo4-xO8)��E(Mn4O8) and E(Co4O8) represent the total energy of per formula unit of MnxCo4-xO8, Mn4O8 and Co4O8, respectively.
According to Table 1, both the total energy and formation enthalpy suggest that Co substitution can improve stability of the structure. It has a consistent declining trend of the total energy; however, the formation enthalpy may well reflect the nature of the structural stability and it is also in good agreement with the experimental analysis[16].
The energy change is mostly due to the electron transfer that causes the interaction force among the ions when Mn atoms are replaced.
3.2 Bond length and volume
The average bond length of Mn��O tends to decrease from in ��-Mn4O8 to in ��-MnCo3O8 as more Co is substituted for Mn (Table 2). By comparison, the average bond length of Co��O is shorter than Mn��O in all primitive cells. It can be observed that bond length of Co��O remains almost unchanged no matter what the Co content is, being fixed at about 0.191 4 nm. The contraction of Mn��O can certainly help to form a more ��-Mn4-xCoxO8 state. The experiment and X-ray absorption spectroscopic studies have born out such a conclusion[4,17]. Although the Mn��O or Co��O bond length is somewhat smaller than the counterpart of experimental value, such a trend of bond length decrease from ��-Mn4O8 to MnCo3O8 is so consistent that the overall errors can be ignored[18].
Table 2 Total energy and formation enthalpy of structure M4O8 with varying Co dopant

There is also a decline in volume from ��-Mn4O8 to MnCo3O8 in Table 2. The average bond length of Mn��O is constricted to a slight extent of about 0.26%. The volume contractions as well as the decrease of Mn��O bond are desirable factors for achieving a stable ��- Mn4-xCox O8 structure in the light of classic chemistry theory. The shorter Mn��O helps to further bind the molecule together in view of atoms and molecules theory, and it follows necessarily that the Co- doped structure contacts more closely[18]. This has been also confirmed by the experimental result[7].
3.3 Ionicity and bond order
According to the Mulliken population(Table 1), the charges distributed to O and Mn in ��-Mn4O8 are -0.39 and 0.79 respectively; whereas the formal charges of O and Mn in ��-Mn4O8 correspond to -2 and +4. The fact that Mn��O bond has not only characteristic of ionicity but also partial covalence can be inferred. As illustrated in Table 1, the Mulliken charge of Mn varies from 0.79 to 0.83 as Mn is gradually replaced with Co and simultaneously the average charge build-up that encompasses O atom appears with a remarkable increase from -0.39 to -0.49. In the light of the electrostatic equilibrium theory, the more charge accumulation there is around atoms, and the more attractive force of the electron density will exert on the molecule[18]. Therefore, it will give an explanation about why the Co-dopant is capable of enhancing the stability of the structure.
As observed in Table 1, the average bond order for Mn��O goes up with Co substitution content increasing. The approximate increment of bond order from ��-Mn4O8 to MnCo3O8 corresponds to 0.06, which is just responsible for the enhancement of the structure. Though the average bond order of Co��O ranks lower than that of Mn��O in magnitude, it still plays a key role in improving the stability of the material by considering its contribution to lift Mn��O bond order up.
3.4 Total and partial electron density of state
For a typical oxide with octahedrally coordinated transition-metal, d orbitals are not equivalent on the scale of energy due to the crystal field splitting effect[1]
(Fig.2). The two  orbitals are of higher energy and lie above the Fermi level extending over from 0 to 3.5 eV in the DOS curve (Fig.3) and they are the main component parts of antibonding. The two eg* can be easily distinguished from the band structure (Fig.2). The bonding region is mainly composed of the O-p orbital, which is generally intermixed with metal-s, metal-p orbitals and metal-d(Fig.2). The range from -19.5 eV to -1.5 eV is regarded as the bonding region. And the extent from -1.5 eV to 0 has a distinctive mark of non-bonding. The structural stability can essentially be determined by the bonding population. The following parts will elaborate on such correlations between the bonding population and structure.
orbitals are of higher energy and lie above the Fermi level extending over from 0 to 3.5 eV in the DOS curve (Fig.3) and they are the main component parts of antibonding. The two eg* can be easily distinguished from the band structure (Fig.2). The bonding region is mainly composed of the O-p orbital, which is generally intermixed with metal-s, metal-p orbitals and metal-d(Fig.2). The range from -19.5 eV to -1.5 eV is regarded as the bonding region. And the extent from -1.5 eV to 0 has a distinctive mark of non-bonding. The structural stability can essentially be determined by the bonding population. The following parts will elaborate on such correlations between the bonding population and structure.
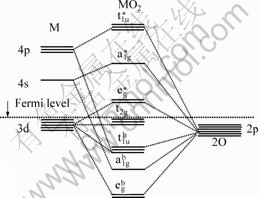
Fig.2 Schematic of band structure expected for oxides with octahedrally coordinated transition-metal cations

Fig.3 DOS and PDOS of primitive cell(Dashed line indicates Fermi level)
The first region involved in bonding part in DOS resides is between -19.5 eV and -17.5 eV and its major part is constituted by metal-s orbital with a very slight metal-d orbital(Figs.2 and 3). It is inevitable that the metal-s and metal-d should only lead to the formation of �� overlap according to the orbital theory[18]. The difficulty is that we can��t decide the intensity of �� overlap because the patterns in DOS along this region keep almost the same. Therefore, we have to examine them carefully by comparing the respective orbital integrals in Table 3. It is evidently seen that the amount of electrons around Mn-s orbital becomes larger with Co dopant increasing and the net gains of electrons on Mn-s amount to 0.04 e from ��-Mn4O8 to MnCo3O8. The difference is likely due to the varying of Co substitution in the material. It is believed that the build-up of electrons along this bonding region contributes to strengthened �� overlap and leads to a more stable structure.
Table 3 Integral of different orbitals indicating total amount of electrons below Fermi level
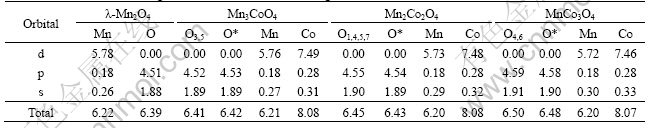
Another region extending from -6.5 eV to -1.5 eV in DOS is a bonding region as well, which consists predominantly of O-p orbital hybridized partially with metal-d orbital(Fig.3). Table 3 shows that the electrons concentrated on the O-p orbital are about 4.51 e in the ��-Mn4O8, and it becomes larger when Mn is continuously replaced with Co, arriving at 4.59 e in the MnCo3O4. Similarly, the charge transfer to O atoms is of great benefit to strongly bind the molecule together.
The DOS peaks around the Fermi level, covering the energy range from -1.5 eV to 0 eV, are mainly made up of the metal-3d orbitals but still partially mixed with the O-p orbital(Fig.3). The bond along this region is characterized as a non-bonding type[1]. It is apparently seen from Fig.3 the DOS peak through the Fermi level shifts down as Mn is replaced. Such a shift, which is probably caused by the lower Co-d states in energy, implies that part of the electrons in the anti-bonding region is transferred to the non-bonding one. As a result, the repulsive force exerted by anti-bonding region will be expected to be weaker, which is easily balanced by the force of attraction from bonding region.
4 Conclusions
1) The mechanism of stability of Co-doped LixMn2O4 (x=0) was investigated by the first-principle plane-wave pseudopotential method. The calculated results show that Co doping plays an important role in modifying the total energy, bond population and electronic structures of ��-MnO2 and results in a more stable structure of Lix Mn2O4 cathode at the end of its charging.
2) The total energy can be reduced considerably and the formation enthalpy is decreased, which indicates that the stability is strengthened due to the Co substitution.
3) Bond length and volume tend to decrease with the content of Co dopant increasing; whereas the bond order of Mn-O increases.
4) The density of states of Mn1- xCoxO2 reveals that more electrons are transferred in the two binding region and the antibonding region eclipses due to the peak of states around Fermi level shift toward the lower energy.
5) Co doping enhances the stability of ��-MnO2 host materials.
Reference
AYDINOL M K, KOHAN A F, CEDER G, et al. Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides[J]. Physical Review B, 1997, 56(3): 1354-1365.
[1] ARORA P, POPOV B N, WHITE R E. Electrochemical investigation of cobalt-doped LiMn2O4 as cathode material for Li-ion batteries[J]. Journal of the Electrochemical Society, 1998, 145(3): 807- 815.
[2] GUMMOW R J, DEKOCK A, THACKERAY M M. Improved capacity retention in rechargeable 4 V lithium/lithium-manganese oxide (Spinel) cells[J]. Solid State Ionics, 1994, 69(1): 59-67.
[3] LIU Yi, FUJIWARAR T, YUKAWA H, et al. Chemical bonding in lithium intercalation compound LixMn2O4(x=0, 1, 2)[J]. Electro- chimica Acta, 2001, 46: 1151-1159.
[4] SHI Si-qi, WANG Ding-sheng, MENG Sheng, et al. First-principles studies of cation-doped spinel LiMn2O4 for lithium ion batteries[J]. Physical Review B, 2003, 67: 115130-115136.
[5] GUOHUA L, IKUTA H, UCHIDA R, et al. The spinel phases LiMyMn2-yO4 (M=Co,Cr,Ni) as the cathode for rechargeable lithium batteries[J]. Journal of the Electrochemical Society, 1996, 143(1): 178-182.
[6] BANOV B, TODOROV Y, TRIFONOVA A. LiMn2-xCoxO4 cathode with enhanced cycle ability[J]. Journal of Power Sources, 1997, 68: 578-581.
[7] AMINE K, TUKAMOTO H, YASUDA H, et al. Preparation and electrochemical investigation of LiMn2-xMexO4 (Me?Ni, Fe, and x=0.5, 1) cathode materials for secondary lithium batteries[J]. Journal of Power Sources, 1997, 68: 604-608.
[8] MIURA K, YAMADA A, TANAKA M. Electric states of spinel LixMn2O4 as a cathode of the rechargeable battery[J]. Electrochimica Acta, 1996, 41: 249-256.
[9] CEDER G, MISHRA S K. Structural stability of manganese oxides[J]. Physical Review B, 1999, 59: 6120-6130.
[10] VANDERBILT D. Soft self-consistent pseudopotentials in generalized eigenvalue formalism[J]. Physical Review B, 1990, 41: 7892-7895.
[11] MATTSSON A E, SCHULTZ P A, DESJARLAIS M P, et al. Designing meaningful density functional theory calculations in materials science��a primer[J]. Modeling and Simulation in Materials Science and Engineering, 2005, 13: 1-31.
[12] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13: 5188-5192.
[13] LIU W, KOWAL K, FARRINGTON G C. Electrochemical characteristics of spinel phase LiMn2O4-based cathode materials prepared by the Pechini process[J]. Journal of the Electrochemical Society, 1996, 143(11): 3590-3596.
[14] STROBEL P, PALOS A I, ANNE M, et al. Structural, magnetic and lithium insertion properties of spinel-type Li2Mn3MO8 oxides (M~Mg, Co, Ni, Cu)[J]. Journal of Materials Chemistry, 2000, 10: 429-436.
[15] WOLVERTON C. First-principles prediction of vacancy order disorder and intercalation battery voltages LixCoO2[J]. Physical Review Letters, 1998, 81(3): 606-609.
[16] HUANG Yu-dai, LI Juan, JIA Dian-zeng. Preparation and characterization of LiMn2-yCoyO4 spinel by low heating solid state coordination method[J]. Journal of Colloid and Interface Science, 2005, 286: 263�C267.
[17] BADER R W. An Introduction to the Electronic Structure of Atoms and Molecules[M]. Oxford: Oxford Press, 1980.
Foundation item: Project(20376086) supported by National Natural Science Foundation of China
Received date: 2006-05-21; Accepted date: 2006-07-27
Corresponding author: HUANG Ke-long, Professor; Tel: +86-731-8879850; E-mail: klhuang@mail.csu.edu.cn
(Edited by YANG Bing)

