


Trans. Nonferrous Met. Soc. China 22(2012) 1717-1722
First-principles study of intrinsic defects, dopants and dopant-defect complexes in LiBH4
ZHANG Guo-ying1, LIU Gui-li2, ZHANG Hui1
1. College of Physics Science and Technology, Shenyang Normal University, Shenyang 110034, China;
2. College of Constructional Engineering, Shenyang University of Technology, Shenyang 110023, China
Received 5 August 2011; accepted 9 November 2011
Abstract: A first-principles study was reported based on density functional theory of hydrogen vacancy, metal dopants, metal dopant-vacancy complex in LiBH4, a promising material for hydrogen storage. The formation of H vacancy and metal doping in LiBH4 is difficult, and their concentrations are low. The presence of one kind of defect is helpful to the formation of other kind of defect. Based on the analysis of electronic structure, the improvement of the dehydrogenating kinetics of LiBH4 by metal catalysts is due to the weaker bonding of B―H and the new metal-like system, which makes H atom diffuse easily; H vacancy accounts for a trace amount of BH3 release during the decomposing process of LiBH4; metal dopant weakens the strength of B―H bonds, which reduces the dehydriding temperature of LiBH4. The roles of metal and vacancy in the metal dopant-vacancy complex can be added in LiBH4 system.
Key words: LiBH4; hydrogen storage material; first-principles calculation; defect; H diffusion; dehydrogenating properties
1 Introduction
Recently, there has been increased focus on sustainable and renewable energy sources as alternatives to the present use of fossil fuels. Among these energy sources, hydrogen is considered an interesting energy carrier in the future, but the key challenge of safe and efficient on-board hydrogen storage has to be solved before the proposed hydrogen society can become a reality. So for realizing hydrogen energy society, we have to develop high-performance hydrogen storage materials.
A potentially interesting material is the complex metal hydride LiBH4, which contains extremely high hydrogen content, 18.4% by mass [1], and partial decomposition of LiBH4 yields 13.5% (mass fraction) of hydrogen through reaction (1) [2].
LiBH4→LiH+B+3/2H2 (1)
However, on-board application of LiBH4 as a successful solid state reversible hydrogen storage material suffers from problems such as a high operating temperatures (above 370 ℃ under a hydrogen pressure of 0.1 MPa), slow kinetics and irreversibility after dehydrogenation due to the chemical inertness of boron [3]. One important approach is to develop a multi component reactive system by incorporating LiBH4 with elemental metals [4,5] or metal hydrides [5,6]. For example, the onset dehydrogenation temperature for the 2LiBH4-Al and 2LiBH4-MgH2 systems is reduced to around 593 K [5] because of the change in reaction pathway described by reactions (2) and (3).
2LiBH4+Al 2LiH+AlB2+3H2 (2)
2LiH+AlB2+3H2 (2)
2LiBH4+MgH22LiH+MgB2+4H2 (3)
The formation of AlB2 or MgB2, instead of pure boron, stabilizes the dehydrogenated state and results in a decrease in ?H for reaction (2) or (3) relative to reaction (1), and is also beneficial to the regeneration of LiBH4. Additionally, a rather high reversible storage capacity is also demonstrated. But the de-/rehydrogenation rates for reactions (2) and (3) are still slow at temperatures below 673 K [5,7]. Another important approach for improving the storage properties of LiBH4 is to incorporate LiBH4 with metal halides. AU et al [8] reported that metal halides TiCl3, TiF3, and ZnF2 effectively reduced the dehydriding temperature through a cation exchange interaction, and some of the halide doped LiBH4 is partially reversible. FANG et al [9] found that TiF3 exhibits a superior promoting effect of TiCl3 on the reversible dehydrogenation of LiBH4. However, even with the aid of these technologies, the dehydriding/ rehydriding properties of LiBH4 are still far below those required for practical application.
To solve problems mentioned above, it is helpful to understand the mechanisms of the reactions of LiBH4 with elemental metals, metal hydrides or metal halides. It is well known that chemical reactions must involve mass transport through bulk crystalline materials, so Li, H and B species’ diffusion in LiBH4 is important in hydrogen uptake and release processes. Mass transport can take place only via lattice defects. For example, hydrogen diffusion [10] was found to be vacancy mediated in NaAlH4, therefore hydrogen-related defect centers, metal impurity dopants and dopant-defect complexes must play a central role in mass transport processes of LiBH4. In the present work, the influence of H vacancy, metal impurity, impurity-vacancy complex on H atoms diffusion and adsorption properties of LiBH4 systems is studied. Some new results will be beneficial to design the advance Li-B-H based hydrogen storage materials.
2 Computation method and models
LiBH4 at ambient conditions is an orthorhombic phase with pnma symmetry (24 atoms/unit cell). We first carried out calculations for bulk LiBH4 to extract basic information about the systems. The calculated lattice parameters are a=7.2713 ?, b=4.4604 ?, and c=6.6212 ? for orthorhombic LiBH4, and the experimental values are a=7.1786 ?, b=4.4369 ?, and c=6.8032 ? [11]. The calculations of native point defects and metal dopants are performed in a (1×1×2) supercell containing 48 atoms (see Fig. 1). We checked the effect of cell size on the formation of HV (hydrogen vacancy), M (metal dopant) and M+HV (metal dopant-hydrogen vacancy complex). With a (1×2×2) supercell, we obtained a 0.015, 0.16, 0.067 eV lower formation energy than that with the (1×1×2) supercell (as calculated with Eq. (5) below), which places a tolerable error bar on the formation energies given below.
All the calculations were performed in the framework of density functional theory (DFT) using generalized gradient approximation (GGA) in the version of Perdew-Burke-Ernzerhof [12] and plane-wave pseudopotential method as implemented in the code CASTEP [13]. Ultrasoft pseudopotential represented in the reciprocal space [14] was used for all elements in our models. All the calculations were carried out using a 3×4×2 Monkhorst-Pack k-point mesh, and the kinetic energy cutoff for the plane wave expansion was set to 270 eV for bulk LiBH4, hydrogen-related defects and dopants, except Ti dopant (set to 280 eV). The convergence with respect to self consistent iterations was assumed when the total energy difference between cycles was less than 1.0×10-6 eV. The geometry relaxation tolerance was better than 0.03 eV/? for force, 0.05 GPa for stress, and 1.0×10-3 ? for displacement.
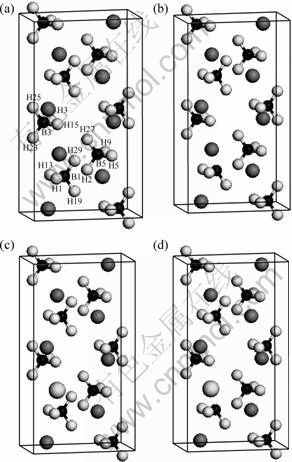
Fig. 1 Models of LiBH4 (a), LiBH4+HV (b), LiBH4+M (c) and LiBH4 + M+ HV (d) systems (Hv denotes vacancy; M stands for metal dapant Ti, Ni, Zr; large gray spheres are M atoms; large dark gray spheres are Li; small dark spheres are B, and small white spheres are H. Some H and B atoms to be used for discussion are labeled as H1, H3, etc and B1, B3, etc)
One of the most common means of atomic diffusion in crystalline solids is via the vacancy mechanism. Conceptually, it can be broken down into two separate processes: 1) vacancy formation and 2) vacancy-atom exchange.
The formation energy of defect is a crucial factor in determining defect concentration. In thermodynamic equilibrium, the concentration c(X) of a defect X at temperature T is given by [15]:
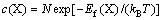 (4)
(4)
where N is the number of sites per unit volume at which the defect can be incorporated; Ef(X) is the formation energy of defect X; kB is Boltzmann constant.
For vacancy diffusion mechanism, vacancy-atom exchange, atom needs to jump into a vacancy site. So atom must break the bonds and squeeze through its neighbors. So we think that the probability of vacancy-atom exchange depends on the bond strength of atom pair near the vacancy, and the stronger the bond strength is, the larger the probability is.
3 Results and discussion
3.1 Defect formation energy
The formation energy of a defect (HV, M or M+HV complex) in LiBH4 is defined as [16]:
Ef(X)=Etot(X)-Etot(bulk)-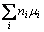 (5)
(5)
where Etot(X) is the total energy of a supercell with a defect; Etot (bulk) is the total energy of the equivalent defect-free supercell; μi is the chemical potential of species i (host atoms or impurity atoms); ni denotes the number of atoms of species i that have been added (ni>0) or removed (ni<0). The chemical potential of hydrogen μH is fixed at Etot(H2)/2. Etot(H2) is determined by the optimization of a 1 nm cube with H2 at eight corners and the bond length of H2 is estimated as 0.0749 nm. The chemical potential of the dopant is fixed by the energy of the involved elemental solid.
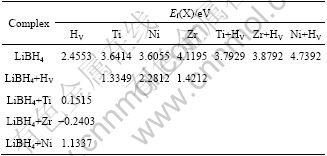
The formation energies of HV, M and M+HV complex are listed in Table 1. From Table1, it can be found that HV and M all have high formation energies, the formation energy of HV decreases greatly in the presence of metal dopant, and the formation energy of M also decreases largely in the presence of HV. The defect formation energy Ef(X) can be used to calculate the defect concentration via reaction (1), so the absolute concentrations of HV and M are quite low, but once one kind of defect (HV or M) has been created, the other kind defect (M or HV) will be created easily, and therefore the defect concentration in doped or defected LiBH4 will be increased largely. For example, HV is with lower formation energy in doped LiBH4. Experimentally, additives such as TiO2, TiCl3 and ZrO2, was found to effectively reduce the dehydrogenation temperature and improve the reversibility of LiBH4. So our calculations are consistent with the experimental observation. Defects interact with one anther and form complexes, such as M+HV complexes. The complexes also have larger formation energies (see Table 1), thus their concentrations are low.
Table 1 Formation energies of defects in LiBH4
3.2 Total density of states
The dehydriding/rehydriding processes of LiBH4 involve hydrogen diffusion, and hydrogen diffusion is vacancy mediated. For vacancy diffusion mechanism, to jump into a vacancy site, H atom needs to possess enough energy (thermal energy) to break the B―H bonds, so hydrogen diffusion is closely relative to the bonding behavior of LiBH4. In this section, we analyze the bonding behavior of LiBH4 by the total density of states (TDOS), and in the next section, we will analyze the bonding behavior of LiBH4 numerically by mulliken overlap population.
The calculations for bulk LiBH4 was first carried out to extract basic information about this system. Figure 2 shows the total density of states (TDOS) for LiBH4. The result for the TDOS of bulk LiBH4 is in qualitative agreement with that reported in previous work [16]. The calculated band gap of LiBH4 is 6.7 eV. This value is comparable to that obtained by MIWA et al [17], namely, 6.8 eV. To our knowledge experimental information about the band gap of LiBH4 is not yet available. It is well known that DFT-LDA (local density approximation) or DFT-GGA produces band gaps significantly smaller than the experimental result. So, the experimental value of LiBH4 band gap should be larger than 6.7 eV. The hydrogen diffusion is vacancy mediated, the total density of states for LiBH4+HV system LiBH4 with H vacancy was calculated and is shown in Fig. 2 also. It is obvious that there is a defect level created by HV at just the middle of the gap, and the Fermi level shifts to the middle of the gap. As a result, the behavior of LiBH4+HV system likes to be a semiconductor, while the perfect LiBH4 is an insulator. This suggests that HV like an electron acceptor pulling electrons from neighbor B―H bond, which leads to changing the distribution of the electrons, and therefore the bond strength of B―H bond near HV and maybe the diffusion of H atom near HV.
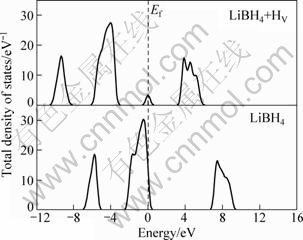
Fig. 2 Total densities of states of LiBH4 and LiBH4 + HV systems (HV denotes vacancy)
In LiBH4, metal additives, such as TiO2, TiCl3 and ZrO2 were found to effectively reduce the dehydrogenation temperature of the system [8]. In order to understand the influence of metal additives on the dehydriding/rehydriding processes of LiBH4, the calculations for LiBH4+M systems (LiBH4 systems with metal dopant) or LiBH4+M+HV (LiBH4 systems with metal dopant+H vacancy complex) were carried out. Figure 3 shows the calculated TDOS for LiBH4+M system (M=Ti, Zr, Ni). It can been clearly seen that metal dopants introduce the impurity levels in band gap, the ones induced by Ti and Zr are shallow levels below the bottom of conduction band, while the one induced by Ni is deep level near the middle of the band gap. Ti, Zr and Ni all make the height of binding peak descend (first peak below the Fermi energy), which suggests that the bonding of Li―B or B―H is weakened. The Fermi energy (Ef) level of LiBH4 +Ti (or Zr) system moves into the conduction band, which indicates that the new system of LiBH4 +Ti (or Zr) has the metal-like behavior. The Ef of LiBH4+Ni system nearly in the middle of the band gap shows that LiBH4+Ni system has the semiconductor-like behavior. Therefore, from electronic structure point of view, one explanation of the improvement of the dehydrogenating kinetics of LiBH4 by metal catalysts is due to the weaker bonding of B―H in the new system, which makes H atom diffuse easily. We can assume that if any substitution element can reduce the bond strength of B―H and make the system become metal-like, it is most likely a candidate element to improve the dehydrogenating kinetics of LiBH4.
Figure 4 shows the total density of states (TDOS) of LiBH4+M+HV (M=Ti,Zr,Ni) systems. In order to see clearly the contribution to defect levels, the local densities of states (LDOS, red lines) of metal atoms are given in Fig. 4. It is clearly seen that there are two kinds of defect levels in the band gap in the presence of dopant-vacancy complex. Compared Fig. 4 with Fig. 3, it can be seen that for Ti+HV and Zr+HV complexes, defect levels introduced by Ti and Zr are below the bottom of conduction band and near Fermi level, and HV introduces the defect levels near the middle of the band gap. For Ni+HV complex, the level created by Ni is just below the middle of the band gap, while the one created by HV is just above the middle of the band gap. Fermi levels move into the conduction band for LiBH4+Ti+HV and LiBH4+Zr+HV systems, but the one of LiBH4+Ni+HV system is in the middle of the band gap. In brief, M+HV complex introduces double defect levels. According to the discussions in the previous section, HV and M all change the distribution of the electrons, and then change the bond strength of B―H, therefore the presence of M+HV complex will trigger a significant enhancement in kinetics and hence in dehydrogenation rate of LiBH4 store hydrogen material.
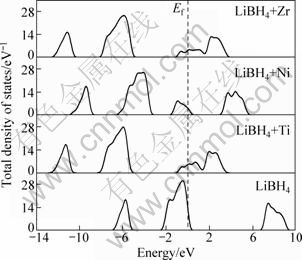
Fig. 3 Total densities of states of LiBH4+M (M=Ti,Ni,Zr) systems
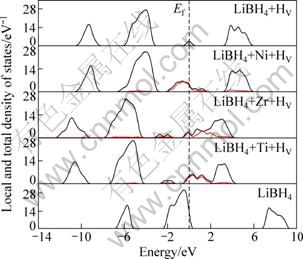
Fig. 4 Local and total densities of states of LiBH4+M+HV (M=Ti,Zr,Ni) systems
3.3 Mulliken overlap population
To further understand the influence of vacancy, impurity, impurity+HV complex on H diffusion, the mulliken overlap population was calculated to analyze the bonding characteristics numerically. Table 2 summarizes the results of the average overlap population. It can be seen that the average population between B―H of perfect LiBH4 is about 0.853 unit bond length (nm-1), indicating the bonding between B―H is strongly covalent.
For LiBH4 with a H29 vacancy, namely LiBH4+HV system, the average populations between B1-H1, B1-H13, B1-H19 are 0.890, 0.890, 0.904 respectively, which are larger compared with that of LiBH4. The creation of a neutral hydrogen vacancy by removing one hydrogen atom from a BH4 unit turns this tetrahedral complex into a trigonal planar BH3 unit (essentially a BH3 molecule). AU and JURGENSEN [18] found that a trace amount of BH3 was released during the decomposing process of LiBH4. Our calculated mulliken overlap population result shows that B―H bonding in BH3 unit gets stronger. So it is suggested that BH3 units in LiBH4+HV system could combine and form B2H6 molecule which may be emitted during the decomposing process of LiBH4. Form Table 2, it can be seen that the B5-H21 bond of the BH4 unit at the right side of hydrogen vacancy (BH3 unit) is weak (the average population is 0.847), so H21 atom can jump to the site of hydrogen vacancy, and therefore H diffusion realizes.
Table 2 Mulliken average overlap population analysis for LiBH4, LiBH4+M, LiBH4+HV and LiBH4+M+HV
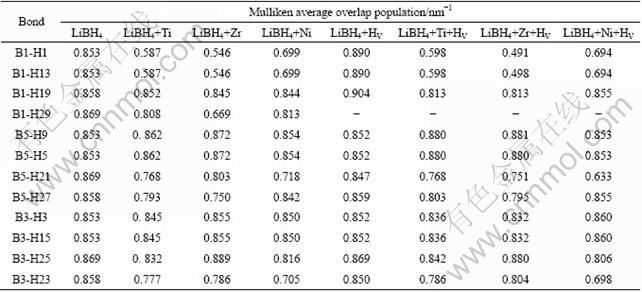
For LiBH4+M systems (see Fig. 1(c)), the strength of B―H bonds (B1-H13, B1-H29, B1-H13, B3-H23, B5-H27) near M are all weaker than those of the corresponding bonds in LiBH4, which reduces the dehydriding temperature of LiBH4 with metals or metal halides.
The average populations between B1-H1, B1-H13, B1-H19 of LiBH4+M+HV are less than those of LiBH4 or LiBH4+HV, which indicates that B2H6 molecule can not form, and B5-H21 bond is also weaker, which is similar to the case in LiBH4+HV. So, we can get the conclusion that in the presence of M+HV complexes, the dehydriding temperature of LiBH4 can be decreased, and the H atom can diffuse easily. From Table 2, we can see that the strength of B―H bonds in LiBH4+Ni or LiBH4+Ni+HV is weaker than that in other metal doped LiBH4 system. This may explain why some metal additives are more effective than others. By analysis above, we found that the roles of M and HV in the M+HV complex can be added in LiBH4+M+HV system.
4 Conclusions
The first-principles plane-wave pseudopotential method based on density functional theory was used to study HV, M, M+HV complex in LiBH4. The formation of HV and M in LiBH4 is difficult, and their concentrations are low. The presence of one kind of defect is helpful to the formation of other kind of defect. Based on the analysis of electronic structure, the improvement of the dehydrogenating kinetics of LiBH4 by metal catalysts is due to the weaker bonding of B―H and the new system metal-like, which makes H atom diffuse easily. In addition, based on the mulliken overlap population analysis, it is found that HV accounts for a trace amount of BH3 release during the decomposing process of LiBH4. Metal dopant weakens the strength of B―H bonds, which reduces the dehydriding temperature of LiBH4. The roles of metal and vacancy in the M+HV complex can be added in LiBH4+M+HV system.
References
[1] SCHLAPBACH L, Z?TTEL A. Hydrogen-storage materials for mobile applications [J]. Nature, 2001, 414: 353-358
[2] Z?TTEL A, WENGER P, RENTSCH S, SUNDAN P, MAURON P, EMMENEGGER C. LiBH4, a new hydrogen storage material [J]. J Power Sources, 2003, 118(1-2): 1-7.
[3] MAURON P, BUCHTER F, FRIEDRICHS O, REMHOF A, BIELMANN M, ZWICKY C N, Z?TTEL A. Stability and reversibility of LiBH4 [J]. J Phys Chem B, 2008, 112: 906-910.
[4] KIM J W, FRIEDRICHS O, AHN J P, KIM D H, KIM S C, REMHOF A, CHUNG H S, LEE J, SHIM J H, CHO Y W, ZUTTEL A, OH K H. Microstructural change of 2LiBH4/Al with hydrogen sorption cycling: Separation of Al and B [J]. Script Mater, 2009, 60(12): 1089-1092.
[5] YANG J, SUDIK A, WOLVERTON C. Destabilizing LiBH4 with a metal (M ) Mg, Al, Ti, V, Cr, or Sc) or metal hydride (MH2 = MgH2, TiH2, or CaH2) [J]. J Phys Chem C, 2007, 111: 19134-19140.
[6] KOU H Q, XIAO X Z, CHEN L X, LI S Q, WANG Q D. Formation mechanism of MgB2 in 2LiBH4 + MgH2 system for reversible hydrogen storage[J]. Transactions of Nonferrous Metals Society of China, 2011, 21(5): 1040-1046.
[7] VAJO J J, SKEITH S L, MERTENS F. Reversible storage of hydrogen in destabilized LiBH4 [J]. J Phys Chem B, 2005, 109(9), 3719-3722.
[8] AU M, JURGENSEN A R., SPENCER W A, ANTON D L, PINKERTON F E, HWANG S J, KIM C, BOWMAN R C Jr. The stability and reversibility of lithium borohydrides doped by metal halides and hydrides [J]. J Phys Chem C, 2008, 112: 18661-18671.
[9] FANG Z Z, KANG X D, YANG Z X, WALKER G S, WANG P. Combined effects of functional cation and anion on the reversible dehydrogenation of LiBH4 [J]. J Phys Chem C, 2011, 115(23): 11839-11845.
[10] SHI Q, VOSS J, JACOBSEN H S, LEFMANN K, ZAMPONI M, VEGGE T. Point defect dynamics in sodium aluminum hydrides―A combined quasielastic neutron scattering and density functional theory study [J]. J Alloys Comp, 2007, 446-447: 469-473.
[11] SOULIE J P, RENAUDIN G, CERNY R, YVON K. Lithium borohydride LiBH4: I. Crystal structure [J]. J Alloys and Comp, 2002, 346(1-2): 200-205.
[12] MARLO M, MILMAN V. Density functional study of bulk and surface properties of titanium nitride using different exchange correlation function [J]. Phys Rev B, 2000, 62(4): 2899-2907.
[13] SEGALL M D, LINDAN P L D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[14] VANDERBILT D. Soft self-consistent pseudo potentials in a generalized eigenvalue formalism [J]. Phys Rev B, 1990, 41(11): 7892-7895.
[15] VAN DE WALLE C G, NEUGEBAUER J. First-principles calculations for defects and impurities: applications to III-nitrides [J]. J Appl Phys, 2004, 95(8): 3851-3879.
[16] HOANG K, van de WALLE C G, NEUGEBAUER J. Hydrogen-related defects and the role of metal additives in the kinetics of complex hydrides: A first-principles study [J]. Phys Rev B, 2009, 80(21): 214109-1-214109-10.
[17] MIWA K, OHBA N, TOWATA S I, NAKAMORI Y, ORIMO S I. First-principles study on lithium borohydride LiBH4 [J]. Phys Rev B, 2004, 69(24): 245120-1-245120-8.
[18] AU M, JURGENSEN A. Modified lithium borohydrides for reversible hydrogen storage [J]. J Phys Chem B, 2006, 110(13): 7062-7067.
LiBH4中本征缺陷、掺杂剂、掺杂剂-缺陷复合体的第一原理研究
张国英1,刘贵立2,张 辉1
1. 沈阳师范大学 物理科学与技术学院,沈阳 110034;
2. 沈阳工业大学 建筑工程学院,沈阳 110023
摘 要:基于密度泛函理论对储氢材料LiBH4中氢空位、金属掺杂、金属掺杂-空位复合体进行第一原理研究。研究发现氢空位和金属掺杂都不容易实现,因此它们的浓度都很低。一类缺陷的存在有助于另一类缺陷的形成。基于电子结构的分析,得出LiBH4中加入金属催化剂导致的释氢动力学性能改善是由于金属催化剂使B―H键减弱,使新体系更具有金属性,从而使氢扩散更容易;氢空位是LiBH4分解过程中释放少量BH3的原因;金属掺杂减弱了B―H键,致使LiBH4的释氢温度降低;在掺杂-空位复合体中,金属掺杂剂和空位的作用可以叠加。
关键词:LiBH4;储氢材料;第一原理计算;缺陷;H扩散;释氢性质
(Edited by YANG Hua)
Foundation item: Project (2009AA05Z105) supported by the High-tech Research and Development Program of China; Project (20102173) supported by the Natural Science Foundation of Liaoning Province, China
Corresponding author: ZHANG Guo-ying; Tel: +86-24-86593291; Fax: +86-24-86575015; E-mail: gyzhang1965@sina.com
DOI: 10.1016/S1003-6326(11)61378-2

