Ni-Mo四近邻作用能对Ni75Al14Mo11合金
沉淀行为影响的微观相场
赵宇宏,侯华,任娟娜
(中北大学 材料科学与工程学院,山西 太原,030051)
摘要:基于微观扩散方程,采用微观离散格点相场法研究Ni-Mo原子间四近邻相互作用能对Ni75Al14Mo11合金沉淀过程微观机制的影响。通过原子尺度的结构演化图、表征浓度和有序度的成分序参数和长程序参数分析沉淀相的有序化、簇聚、镍原子反向析出及粗化行为等。研究结果表明:最近邻、第三近邻原子间作用能增大,可促进沉淀相的簇聚及有序化,但抑制后期镍基原子团簇的反向析出及粗化;次近邻、第四近邻原子间作用能增大的影响则与之相反;在相同条件下,外层作用能对沉淀相的有序化和簇聚影响最大。
关键词:微观相场法;四近邻作用能;Ni75Al14Mo11合金;沉淀机制
中图分类号:TG146.2+1 文献标志码:A 文章编号:1672-7207(2012)08-2964-09
Microscopic phase-field simulation for influence of Ni-Mo fourth-nearest interaction energy on precipitation process
of Ni75Al14Mo11 alloy
ZHAO Yu-hong, HOU Hua, REN Juan-na
(College of Materials Science and Technology, North University of China, Taiyuan 030051, China)
Abstract: The influence of Ni-Mo fourth-nearest atomic interaction energy on the precipitation mechanism of Ni75Al14Mo11 alloy was investigated with the phase field model based on the microscopic diffusion equation. The structure evolution on atomic scale, the composition and long-range order parameters of the precipitates were analyzed to explore the ordering and clustering, atoms reverse precipitation and coarsening occurred in precipitation process of Ni75Al14Mo11 alloy. The results show that the increase of the nearest/the third Ni-Mo atomic interaction energy can promote the atoms clustering and ordering, yet restrain the later Nickel atoms reverse precipitation and coarsening; the influence of the second/the fourth Ni-Mo atomic interaction energy is contrary to that of the nearest/the third; and the influence of the outermost atomic interaction energy on clustering and ordering is the largest in the same condition.
Key words: microscopic phase field model; fourth-nearest interaction energy; Ni75Al14Mo11 alloy; precipitation process
原子间作用能对微观结构的影响目前备受关注,主要原因是:一、原子间结合力的能力决定了材料的结构、电磁特性、内禀力学,对材料的性能有重要影响;二、电场和磁场在我们生活中随处可见,而电场和磁场作用在合金的沉淀过程中是以原子间作用能的变化形式出现的,即合金在发生沉淀时,如果有电场或磁场出现,那么合金中的原子间作用能将发生变化,所以研究原子间作用能变化可间接地研究外场(电场、磁场等)对合金沉淀的影响。Ni-Al-Mo合金在沉淀过程中析出的Ni3Al有序相为强化相,对合金的机械强度、韧性、抗蠕变和抗磨损等性能具有重要的影响。在Ni-Al-Mo体系中考虑3种原子的跃迁及可能的排列,采用原子间四近邻相互作用近似可更准确地描述体系自由能[1]。Clouet等[2]在Al-Zr及Al-Sc原子间相互作用势基础上,利用Monte Carlo方法模拟了Al3Zr和Al3Sc的形核机制。Vailhe等[3]利用嵌入原子模型模拟了Ni53Al35Fe12合金的原子间相互作用势及位错。陈铮等[4-7]模拟了不同合金体系在不同原子间相互作用势下的沉淀行为,如有序化、沉淀机制、原子的替代、孕育期等。但以上研究并没有考虑原子间作用能改变对沉淀相的有序化、簇聚和基体原子反向析出、粗化的影响。相场法中相场方程的建立是基于热力学驱动力和有序化势的共同作用,因此相场法可以通过描述一系列浓度、长程序参数等场变量,来描述微观组织结构及其演化过程。相场法假设相与相之间的界面为连续的,与经典理论模型中假设的明锐界面不同,更接近实际,因为在相和相之间是存在一个过渡区域的,而这个过渡区域的厚度与核心的尺寸相比是不可忽略的,所以为了能够更加接近问题的真实值,相场法越来越受到人们的青睐。本文作者采用三元微观相场模型,通过研究Ni-Mo原子间四近邻作用能的变化,对Ni75Al14Mo11合金沉淀过程进行分析,得到四近邻原子间作用能对沉淀过程前期Ni3Al相的有序化、簇聚和后期镍基原子反向析出及粗化的影响规律。
1 理论模型
1.1 微观相场动力学方程
相场动力学方程基于Onsager和Ginzburg-Landau[8-9]理论,用原子占位几率描述扩散和沉淀过程。对于三元体系,用PA(r,t),PB(r,t),PC(r,t)分别表示A,B,C原子在 t时刻、占据格点位置r的概率,并添加随机项来模拟热起伏,得到Langevin微观相场方程为:
 (1)
(1)
式中:T为热力学温度;kB为波尔兹曼常数; 为与单位时间内一对A和B原子在格点位置r和r′上的交换概率有关的常数;F为系统的总自由能;t为时效时间步数;ξ(r,t)是均值为0的高斯分布。
为与单位时间内一对A和B原子在格点位置r和r′上的交换概率有关的常数;F为系统的总自由能;t为时效时间步数;ξ(r,t)是均值为0的高斯分布。
1.2 四近邻原子间相互作用能
系统总自由能F为[10]:
 (2)
(2)
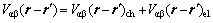 (3)
(3)
其中: 为原子间有效作用能,
为原子间有效作用能,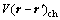 为原子间的化学相互作用能,
为原子间的化学相互作用能, 为原子间的弹性相互作用能,这里,下标α,β分别代表原子A,B或C。为了更准确地描述自由能,采用四近邻原子间相互作用势,
为原子间的弹性相互作用能,这里,下标α,β分别代表原子A,B或C。为了更准确地描述自由能,采用四近邻原子间相互作用势,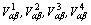 分别是原子α与β的第一、二、三、四近邻原子间相互作用能,在面心立方倒易空间中有:
分别是原子α与β的第一、二、三、四近邻原子间相互作用能,在面心立方倒易空间中有:

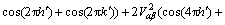
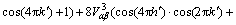
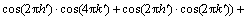
 (4)
(4)
其中,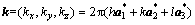 ;h,k和l分别为倒易晶格位置;
;h,k和l分别为倒易晶格位置; ,
,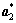 和
和 为倒易晶格单位矢量。
为倒易晶格单位矢量。
在波矢近似为0的长波近似为:
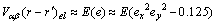 (5)
(5)
式中:E为表征弹性性质和晶格失配的应变能,可表示为:
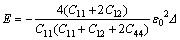 (6)
(6)
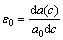 (7)
(7)
式中:Δ=C11-C12-2C44为弹性各向异性常数;C11-C12-2C44是模型体系基体的弹性常数;c为原子的溶质的原子数分数;a(c)为浓度为c时的晶格点阵参数;e为k方向上的单位矢量;ex和ey分别为e在倒易空间中沿x,y轴的分量。
求解式(1)~(5)可得到每个格点位置原子的占位 概率。
1.3 原子有序化及原子簇聚
采用局部长程序参数η(i,j)表示局部区域的原子有序化程度[11]:
 (8)
(8)
式中,p(i,j)为原子在(i,j)位置的占位概率;c(i,j)为原子在晶格中的平均占位概率;当η(i,j)=1时,原子为完全有序状态,当η(i,j)=0时,为对应无序状态。
平均长程序参数可表示整个区域原子有序化的程度,可通过对局部长程序参数η(i,j)取绝对平均得到;平均成分偏离序参数是通过对局部成分序参数c(i,j)与初始值的偏离取绝对值得到,可表示整个区域原子簇聚的程度。
2 模拟结果及分析
基于Ni75Al14Mo11 合金在1 073 K下时效的有效原子间作用能,将Ni-Mo的第一、二、三和四近邻原子间作用能分别±10 meV,研究其改变对该合金沉淀过程中原子有序化和簇聚等行为的影响。
2.1 Ni75Al14Mo11合金沉淀过程的微结构演化
图1所示为1 073 K时Ni75Al14Mo11合金在不同模拟时间上的微结构演化过程。从图1(a)可知:t=17 000时,合金仍处于无序状态,存在少许浓度起伏,少量的Ll2有序结构的Ni3Al沉淀相开始显现,尺寸较小,随机分布,彼此之间相互独立存在,随着时间延长,有序相尺寸一直在长大,并发生碰撞、融合,部分小的颗粒融入邻近大颗粒中,即发生小畴消失、大畴长大的粗化过程,有序相之间形成的界面发生迁移;图1(d)中合金发生分解,溶质Mo原子开始从相界处析出;图1(e)中镍基原子开始从沉淀相中反向析出;图1(f)中发生镍基原子从沉淀相中反向析出及粗化过程,使得有序相间的界面变宽。
2.2 改变四近邻作用能对合金沉淀行为的影响
2.2.1 t=30 000时原子占位概率随原子间相互作用能的变化
改变各近邻原子间作用能时Ni75Al14Mo11合金在t=30 000时的沉淀微结构变化如图2所示。其中,Vi为第i近邻原子间相互作用能,i=1,2,3,4。比较图2(a1)、图2(a2)和图1(c)中沉淀相的有序程度发现:随着最近邻作用能的增大,沉淀相的有序度增加,可见对该合金的有序化起促进作用;同理,分别比较图2和图1发现:第三近邻作用能对沉淀有序化的影响和最近邻的影响一致,增加第三近邻作用能可促进合金的有序化过程;次近邻与第四近邻作用能的影响一致,增加次近邻或第四近邻作用能,对合金的有序化起抑制作用。从图2可见:发现最外层原子间作用能改变对其影响程度最大。
2.2.2 t=200 000时原子占位概率随原子间相互作用能的变化
图3所示为t=200 000时,不同原子间作用能下Ni75Al14Mo11合金的微结构演化图。比较图3(a1),图3(a2),图1(e)发现:图3(a1)反向析出的镍基原子数量比图1(e)中的多,而图1(e)中反向析出的镍基原子数量比图3(a2)中多,可见此时最近邻作用能小的镍基原子反向析出量比最近邻作用能大的镍基原子反向析出量大,即最近邻作用能的增加抑制了镍基原子的反向析出。同理,分别比较图3和图1发现,第三近邻作用能对镍基原子反向析出的影响和最近邻的影响一致,即增加第三近邻作用能抑制了合金在沉淀过程中镍基原子的反向析出;次近邻与第四近邻作用能的影响一致,增加次近邻或第四近邻作用能,对镍基原子的反向析出起促进作用。
2.2.3 t=500 000时原子占位概率随原子间相互作用能的变化
图4所示为t=500 000时,改变原子间作用能时Ni75Al14Mo11合金的沉淀微观结构图。比较图4(a1),图4(a2)和图1(f)发现:最近邻作用能的增加抑制了反向析出的镍基原子团簇的粗化过程。同理,分别比较后面3组可知:次近邻、第四近邻作用能的增加对反向析出的镍原子团簇粗化过程起促进作用,第三近邻作用能的增加抑制了反向析出镍原子团簇的粗化 过程。
由此可见:Ni-Mo四近邻原子间作用能对沉淀过程的前期和后期影响是不同的,随着最近邻、第三近邻原子间作用能增大,可促进沉淀相的有序化和簇聚,但在镍原子团簇反向析出和粗化的过程中却起抑制作用;次近邻、第四近邻原子间作用能增大,抑制了沉淀相的有序化和簇聚,但在镍基反向析出和粗化的过程中却起了促进作用。Ni-Mo四近邻作用能的增大,且在每层作用能增大同一数值时,外层有序能对沉淀相的有序化和簇聚行为影响最大。
2.3 对沉淀全过程原子簇聚和有序化的影响
(b2) V2+10meV
(b1) V2-10meV
图5所示为改变Ni-Mo四近邻原子间作用能对应的平均序参数随时间变化的影响。从图5可看出:平均序参数(包括平均长程参数和平均成分序参数)在t=10 000以前均为0,此时体系处于沉淀的孕育期,基体中没有新相形成;在t=10 000~30 000,平均序参数迅速上升到较高值,对应的是沉淀相的有序化和簇聚;t=30 000之后,进入平衡阶段,发生粗化过程。
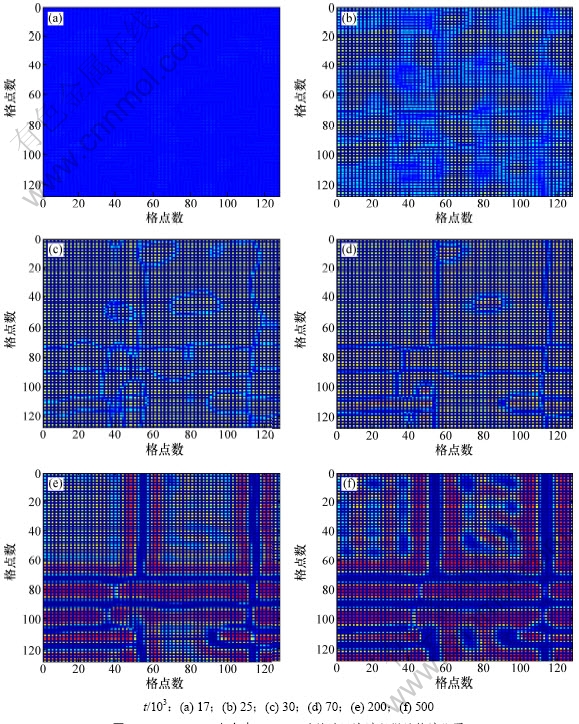
图1 Ni75Al14Mo11合金在1 073 K时效时沉淀过程微结构演化图
Fig.1 Simulated precipitation microstructure evolution of Ni75Al14Mo11 aged at 1 073 K
从图5(a)和图5(b)可见:平均序参数的分布规律从小到大为:V1-10 meV,V1,V1+10 meV,即Ni-Mo最近邻原子间作用能增加对合金沉淀过程的有序化和簇聚起促进作用。同理,从图5(a3)和图5(b3)也可看出:长程序参数和成分序参数从小到大的顺序为:V3-10 meV,V3,V3+10 meV,即Ni-Mo第三近邻原子间作用能增加促进了合金沉淀过程的有序化和簇聚。由图5(a2)和图5(b2)可知:在同一时刻,长程序参数和成分序参数从大到小的顺序为:V2-10 meV,V2,V2+10 meV,说明在同一时刻,Ni-Mo次近邻原子间作用能增加对合金沉淀过程的有序化和簇聚起抑制作用。同理,从图5(a4)和图5(b4)也可看出:Ni-Mo第四近邻原子间作用能增加抑制了合金沉淀过程的有序化和簇聚。

图2 改变各近邻原子间作用能时Ni75Al14Mo11合金在t=30 000时的沉淀微结构变化
Fig.2 Simulated precipitation microstructure evolution of Ni75Al14Mo11 when t=30 000 with changed interaction energy
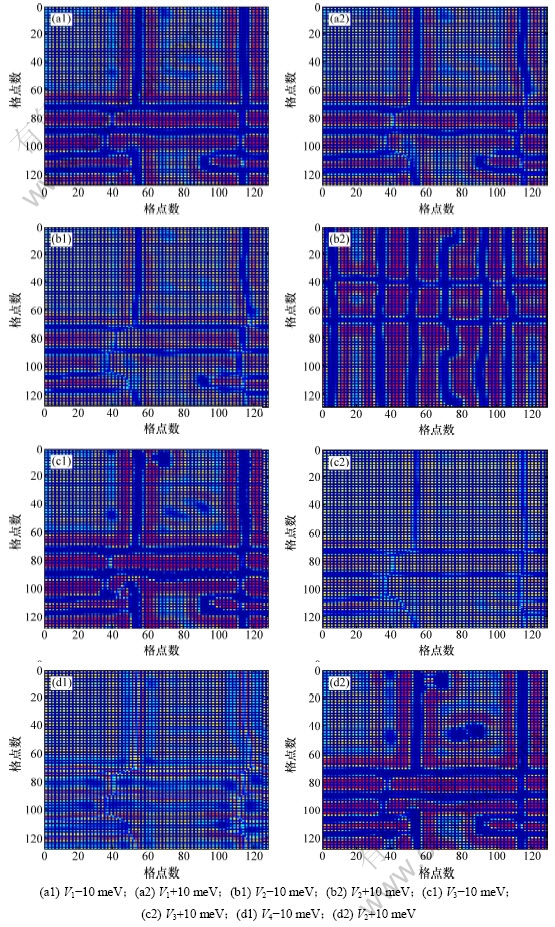
图3 改变各近邻原子间作用能时Ni75Al14Mo11合金在t=200 000时的沉淀微结构变化
Fig.3 Simulated precipitation microstructure evolution of Ni75Al14Mo11 when t=200 000 with changed interaction energy
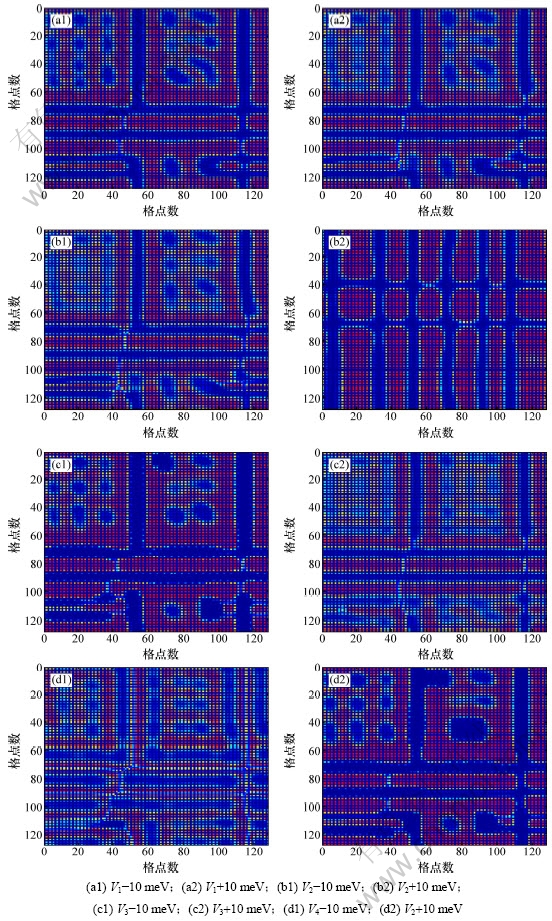
图4 改变各近邻原子间作用能时Ni75Al14Mo11合金在t=500 000时的沉淀微结构变化
Fig.4 Simulated precipitation microstructure evolution of Ni75Al14Mo11 when t=500 000 with changed interaction energy

图5 改变各近邻原子间作用能对Ni75Al14Mo11合金沉淀全过程原子簇聚和有序化的影响
Fig.5 Clustering and ordering during precipitation process of Ni75Al14Mo11 with changed interaction energy
从图5可知:在同一时刻第四近邻作用能的改变对成分偏离序参数的影响最大,即对沉淀相原子簇聚的影响最大;发现第四近邻作用能的改变对沉淀相有序化的影响最大。
3 结论
(1) 第一、三层Ni-Mo原子间作用能增大,促进了Ni75Al14Mo11合金过程中早期沉淀相的有序化和簇聚,抑制了后期镍基反向析出及粗化。
(2) 第二、四层Ni-Mo原子间作用能增大,抑制了Ni75Al14Mo11合金过程中早期沉淀相的有序化和簇聚,促进了后期镍基反向析出及粗化。
(3) 外层Ni-Mo原子间作用能对沉淀的前期的影响较大,内层原子间作用能的影响较小。
参考文献:
[1] Chen L Q. Computer simulation of spinodal decomposition in ternary systems[J]. Acta Metall Mater, 1994, 42(10): 3508-3515.
[2] Clouet E, Nastar M, Sigli C. Nucleation of Al3Zr and Al3Sc in aluminum alloys: From kinetic Monte Carlo simulation to classical theory[J]. Phys Rev B, 2004, 69(6): 064109-064123.
[3] Vailhe C, Farkas D. Trends in dislocation core structures and mechanical behavior in B2 aluminides[J]. Materials Science and Engineering, 1998, 258: 26-31
[4] 赵彦, 陈铮, 王永欣. Ni-Al有序能对Ni75Al13Cr12合金原子有序行为影响的微观相场模拟[J]. 稀有金属材料工程, 2009, 38(10): 1756-1760.
ZHAO Yan, CHEN Zheng, Wang Yong-xin. Microscopic phase field simulation for the influence of Ni-Al ordering energy on ordering behavior of Ni75Al13Cr12 alloy atoms[J]. Rare Metal Materials and Engineering, 2009, 38(10): 1756-1760.
[5] 甑辉辉, 王永欣, 陈铮, 等. 四近邻对势对Ni75Al5V20合金沉淀机制影响的微观相场模拟[J]. 稀有金属材料与工程, 2009, 38(2): 286-290.
ZHEN Hui-hui, WANG Yang-xin, CHEN Zheng, et al. Microscopic phase-field simulation for the effect of the fourth-nearest pair potentials on precipitation mechanism of Ni75Al5V20 alloy[J]. Rare Metal Materials and Engineering, 2009, 38(2): 286-290.
[6] 赵彦, 陈铮, 王永欣, 等. 有序能对Ni75Al15Cr10合金Cr替代行为影响的微观相场研究[J]. 金属学报, 2009, 45(5): 635-640.
ZHAO Yan, CHEN Zheng, WANG Yong-xin, et al. A microscopic phase-field study for the influence of ordering energy on Cr substitution behavior in Ni75Al15Cr10 alloy[J]. Acta Metallurgica Sinica, 2009, 45(5): 635-640.
[7] 徐聪, 陈铮, 王永欣, 等. 原子间相互作用能对Ni75AlxV25-x合金孕育期影响的微观相场模拟[J]. 稀有金属材料与工程, 2009, 38(11): 1935-1939.
XU Cong, CHEN Zheng, WANG Yong-xin, et al. Phase-field simulation for the effect of atomic interchange energy on the incubation time of Ni75AlxV25-x alloy[J]. Rare Metal Materials and Engineering, 2009, 38(11): 1935-1939.
[8] Poduri R, Chen L Q. Computer simulation of the kinetics of order-disorder and phase separation during precipitation of Al3Li in Al-Li alloys[J]. Acta Materialia, 1997, 45(1): 245-255.
[9] Miyazaki T, Koyama T, Kozakai T. Computer simulation of the phase transformation in real alloy systems based on the phase field method[J]. Materials Science and Engineering A, 2001, 312: 38-49.
[10] Poduri R, Chen L Q. Computer simulation of morphological evolution and coarsening of Al3Li precipitates in Al-Li alloys[J]. Acta Materialia, 1998, 46(11): 3915-3928.
[11] Chen L Q, Simmons J A. Microscopic master equation approach to diffusional transformations in inhomogeneous systems-single site approximation and direct exchange mechanism[J]. Acta Metal Mater, 1994, 42(9): 2943-2954.
(编辑 赵俊)
收稿日期:2011-06-25;修回日期:2011-09-21
基金项目:教育部博士点新教师基金资助项目(20101420120005);山西省留办基金资助项目(2010-78);国家自然科学基金资助项目(50975263);科技部国际合作项目(2011DFA50520)
通信作者:赵宇宏(1974-),女,河北石家庄人,博士,从事计算材料学研究;电话:15035172958;E-mail:zyh388@sina.com

