DOI��10.19476/j.ysxb.1004.0609.2017.12.19
La����AgSnO2��ͷ���ϵ������ܵĵ�һ��ԭ������
�Բ��������ۣ�����骣���¶¶���� ٻ
(�ӱ���ҵ��ѧ ��ų�������ɿ���ʡ�������ص�ʵ���ң���� 300130)
ժ Ҫ��AgSnO2��һ�ֽ�Ϊ�����AgCdO������ϣ�����������SnO2������Ե��ʹ�ô�ͷ���ϵĽӴ��������ʸ���SnO2�ĵ������Ǽ��������ش����⡣���û����ܶȷ������۵ĵ�һ��ԭ�����Ʒ���ͨ����ģ�ķ������ֱ�����ͬ����(50%��25%��16.67%��12.5%��8.34%)La���ӵ�SnO2����ģ�ͣ�����������侧��������ɲ��ӡ��ܴ��ṹ��̬�ܶȵ����ʡ����������La���Ӻ�ͷ�����е�SnO2������ֱ�Ӵ�϶�뵼����ϣ�����������ȱ�С��������Ũ�ȱ��ʹ�ò��ϵĵ�������ǿ����La���ӱ�Ϊ16.67%ʱ��������ѡ�
�ؼ��ʣ�La����SnO2����һ��ԭ�������ӽṹ����ѧ����
���±�ţ�1004-0609(2017)-12-2552-08���� ��ͼ����ţ�TM501���� ���ױ�־�룺A
Ŀǰ��AgCdO��ͷ�������ں����ж�����Cd�������˳��г���ȡ����֮����AgSnO2��ͷ���ϡ��ò�����Ҫ���ڽӴ�������·�����̵����ȵ�ѹ�����У���һ��Ag��SnO2�Ļ�������AgΪ��Ҫ�ɷ֣�Ϊ��ֹAg�ܻ���Һ��ɽ�������SnO2��һ������ǿ��Һ�ȵ����ã���SnO2�ز����١�����SnO2����һ�ֿ������뵼����ϣ����ƾ�Ե�����϶���Ⱥܿ����ﵽ3.6 eV[1-2]�����䵼���Լ�����봥ͷ�����ж��䵼����Ӱ�켫��ʹ�ô�ͷ���ϵĽӴ��������������о�����������SnO2�����γɵ�����������ܹ�ʹ�ýӴ������С����������ǿ[3-5]������ѧ����SnO2���ӵĵ�һ��ԭ���о����������˹���[6-8]���ڷ��[9]�õ�һ��ԭ�������о���SnO2������ܴ��ṹ����ѧ�ѵ�[10]��Ru����SnO2�뵼����е��ӽṹ���о�������Ru����SnO2ȷʵ�ܹ���߲��ϵĵ����ԣ���÷��[11]�о���Sb����SnO2���ϵĹ�����ʣ����ý���ָ�������Ӻ�SnO2���ϵ����Եõ�������ǿ�����а���������ʣ������ܼ����ܴ�ϸ���������õ�[12]��Ce����SnO2���Ͻṹ��ѧ���ʽ����о����������Ӻ���Ͼ���ֱ��ԾǨ�뵼�壬Ce��4f������뵼����ʹ�õ�������ܶ��ƶ����������ȱ�С���ֽ�Ǭ��[13]��Ti����SnO2�뵼���������ӽṹ�����ܽ������о����������Ӻ�IJ�����Ϊֱ�Ӵ�϶���������Ų��ӱ��������ӣ���϶��С��������[14]�Թ��ɽ���V��Cr��Mn���ӵ�SnO2��ϵ���ӽṹ�����о�������V��Cr���Ӻ�SnO2���а���������ʣ���Mn���Ӻ�û�з����������ʣ�������[15]�Թ��ɽ���Cr����SnO2�����ܼ������ܽ����о�����������ʹ����ϵ���ְ�����ԡ����ǣ�����La����SnO2�뵼������ۼ��㻹δ������������������Ҫ���û����ܶȷ�������(Density functional theory��DFT)�ĵ�һ��ԭ�����������۽Ƕȼ����˲��Ӻ�ͷ�����е�SnO2�ܴ��ṹ��̬�ܶ�ͼ���о�ϡ��Ԫ��La����SnO2�������ܣ�Ϊ����AgSnO2��ͷ���ϵ������ṩ��һ�����۷�����
1 La����AgSnO2��ͷ������SnO2�ľ���ģ������㷽��
��ͷ�����е�SnO2Ϊ���ʯ�ͽṹ���ռ�ȺΪ136 P4/MNM���������ķ���ϵ�����к���2��Snԭ�Ӻ�4��Oԭ�ӣ�Snԭ���ھ���Ķ�������ġ�������Ϊ��a=b=0.4737 nm��c=0.3816 nm����=��=��=90o����ͼ1��ʾ[16-18]��
��SnO2����ԭ������ķ�����������ͬ�ľ���ģ�ͣ��ٽ����е�һ��Snԭ���滻ΪLaԭ�ӣ�����������ӱ����Ķ�Ӧ��ϵ���1����[19-20]��

ͼ1 SnO2ԭ���ṹ
Fig. 1 Model of primitive cell of SnO2
��1 ���ӱ����볬�����Ķ�Ӧ��ϵ
Table 1 Correspondence relationship between doping ratio and super cell

ͨ���ܶȷ����ĵ�һ��ԭ����Ǣ�����ܹ��ó������ӵIJ��������Ӷ��õ���ϵ�������͵���ܶȷֲ�[21-24]��������̲��ù����ݶȽ��Ʒ�(GGA)������Ԩ������4��4��6��Monkorst-Park������ƽ�沨�ض���ѡ��380 eV���Ż�������BFGS�㷨��ԭ�Ӽ�������������涨Ϊ0.3 eV/nm����Ӧ�����ֵ�趨Ϊ0.1 GPa��ѡ��۵�����̬�ֱ�ΪSn��5s2 5p2��O�� 2s2 2p4��La��5p6 5d1�����������ļ�����ڵ��ռ��н���[25]��
2 La����AgSnO2��ͷ������SnO2�ļ����������
2.1 ����ṹ
���ȶԲ�ͬ���ӱȵľ���ṹ�����Ż����Ż���ľ�����������2���С�
�ɱ����е����ݿ�֪�Ż�ǰ��������ʵ���������ݻ���������ʿ����������һ�����㡣�ӱ����з����õ�����La����֮�ϵͲ��ӱ�ʱ����������Ͳ�������������La�����������Ӷ����ӡ�������ΪLa3+���Ӱ뾶(0.106 nm)��Sn4+���Ӱ뾶(0.083 nm)Ҫ��ö�[26]�������Ӱ뾶���La3+����ȡ�������Ӱ뾶С��Sn4+���ӣ�����La��O���ļ���(0.2417 nm)Ҳ����Sn��O���ļ���(0.2081 nm)���������ӻ�ѧ���۵�֪ʶ������La�������������������������������������ݡ�
����ͬ�ļ��������£������Ż���ģ����Ϊ�������ֱ�����˲�ͬ���ӱ��µĵ����ܡ������ܹ���ӳ�����²��ϵ��ȶ��ԡ��ɱ�2��֪�����Ų��ӱȵ����ӣ��������ڲ��Ͻ��ͣ��ʲ��ϵ��ȶ����ڲ�����ǿ���ڲ��ӱ����50%ʱ����������ͣ���ʱ���ϵ��ȶ�����á�
2.2 Mulliken���ӷ�������ط���
���ڲ�ͬ�IJ��ӱȵļ������һ�µĻ��������ʿɱȽϲ�ͬ���ӱ��µ�ԭ�ӵ�ɷֲ������ԭ�Ӳ��ӡ���3�����˱���SnO2��50%���ӱ��µ�SnO2����ԭ�ӵĵ�ɲ��ӡ�
��2 ʵ���뼸���Ż���������
Table 2 Crystal parameters of geometry optimization and experimentation

��3 SnO2����ӱ�Ϊ50%��SnO2ԭ�ӵ�Mulliken���ӷ���
Table 3 Mulliken population analysis of SnO2 and 50% doped SnO2

�ɱ�3���Կ�����O1��O2ԭ�ӵ���Ч������ɲ���ǰ��-0.95e��Ϊ-0.80e��O3��O4ԭ�ӵ���Ч������ɲ���ǰ��-0.95e��Ϊ-0.78e����Щ�仯��Ҫ������p����ϣ�Sn1ԭ�ӵ���Ч������Լ�La���Sn2ԭ�Ӻ����Ч���������δ����ʱС����Ҫ�仯��s��p����Լ�La��d����ϣ�˵�����Ӻ��Sn1ԭ�Ӻ�Laԭ��ʧ���������٣����Ӻ��Sn��O��La��O֮��������Լ�������������ǿ��
��4��ʾ�˲���ǰ���ԭ�Ӽ������ص����������ɱ������ݿ�֪���ڲ��Ӻ�La�����Oԭ��֮��IJ�������С��δ���ӵ�Sn2�����Oԭ��֮��IJ�����������������˵��������ϵ��La��O���������Դ���Sn��O�������ǿ�ȱ�С����������La������ϵ��������Խϲ����La��ԭ�Ӱ뾶��Sn�Ĵʳɼ��ļ����ϴ�����ϴ�����La�����ʣ�ʹ��������ͷ���������ӣ����ϵĿ��ۺ�����ǿ����Ҳ��������֤ʵ��La�����ܹ����AgҺ����[27-28]������Sn1�����Oԭ��֮��IJ�����������֮���Sn1�����Oԭ�ӵ��ص����������ӣ�������������˵������֮���Sn��O�����ǿ�ȴ���δ���ӵ�Sn��O������������Sn1��Oԭ��֮��������Լ�����������ǿ���£����������Լ����ij̶����СһЩ����ʹ�ù��������ֳ���������ΪSn��O�����ǿ�ȱ���ڼ���������������Laԭ�ӵIJ���ʹ�þ�������������¡�
��4 SnO2����ӱ�Ϊ50%��SnO2ԭ�ӵļ������ص�������
Table 4 Atomic bond length and overlap population of SnO2 and 50% doped SnO2

ͼ2(a)��ʾΪ����SnO2�뵼��ĵ���ܶȷֲ�ͼ��ͼ2(b)��ʾΪ���ӱ�Ϊ50%ʱSnO2�ĵ���ܶȷֲ���ͼ�б�ע�ĺ졢�ơ�������ɫ����ֱ��ʾ����ܶȵĴ�С���Ա�������ܶȷֲ�ͼ��֪��Laԭ����Χ�ĵ���ܶ�����С��Snԭ�ӣ��������Laԭ�ӵ���ܶ��������࣬��������ϡ��Ԫ�ص�f������á����Ӻ�Laԭ����Oԭ����Χ�ĵ������ص��̶ȱ���෴�ģ�Snԭ����Oԭ����Χ�ĵ������ص��̶ȼ�С������Oԭ�ӵĵ���ܶȣ����Ӻ�Oԭ����Χ����ܶȱȲ���ǰС���������������ĵ�ɲ��������Ǻϡ�

ͼ2 ����ǰ�����ܶȷֲ�ͼ
Fig. 2 Distribution of charge density
2.3 �ܴ��ṹ��̬�ܶ�
�ɵ�һ��ԭ���ļ��㷽�����Եõ�������ϵ���ܴ��ṹ��̬�ܶȣ�Ϊ�˷���Աȷ������Ӷ��ڴ�ͷ�����е�SnO2�ĵ��ӽṹ��Ӱ�죬���ȶ�δ����SnO2���ϵ��ܴ�ͼ�Լ����ӱ�Ϊ16.67%��SnO2���ܴ�ͼ�����˼��㣬��������ͼ3��4��ʾ��
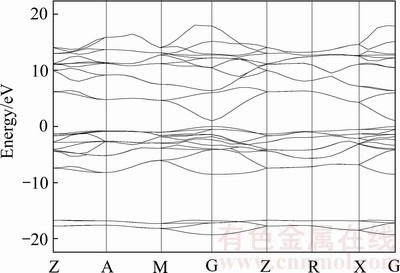
ͼ3 SnO2���ܴ��ṹͼ
Fig. 3 Band structure of SnO2

ͼ4 ���ӱ�Ϊ16.67%ʱSnO2���ܴ��ṹ
Fig. 4 Band structure of 16.67% doped SnO2
ͼ3��ʾΪSnO2���ܴ�ͼ��ͼ4��ʾΪ���ӱ�Ϊ16.67%ʱ��SnO2���ܴ��ṹͼ�����߾�ѡ��0 eV��Ϊ�����ܼ�EF����ͼ3��4���ܹ�������SnO2��������͵���۴�����ߵ���ڲ���Ԩ����G�㣬�ɴ˿�֪SnO2����ֱ�Ӵ�϶�뵼����ϣ����Ҳ���ǰ��SnO2�뵼��ṹ���䡣�����뵼��SnO2���ù����ݶȽ��Ʒ�(GGA)���м���õ��Ĵ�϶ֵΪ1.580 eV������[29]����������[30]�����ô˷�������Ľ��Ϊ1.4��1.258 eV�����о���������֮����һ�¡����ǣ���϶������ֵΪ3.6 eV�����ó��ļ�����ԶԶС��ʵ��ֵ����������ʹ��Castep���������ձ���ڴ�϶ֵ����ƫ�͵����⡣�����ܶȷ��������е�KS���̵ı���ֵ����ȷ�ظ���ϵͳ�ļ���������ʹ��λ�ڵ����ĵ���̬����ֵ��ʵ��ֵƫ�ͣ�����ó��Ĵ�϶ֵ���ڻ�̬״̬�£���ʵ��õ��Ĵ�϶ֵ���ڼ���̬״̬�£����Ծͻ���ɼ�������[31-32]��BERGER��[33]����GW�㷨�ܹ��õ�SnO2�Ĵ�϶ֵΪ3.5 eV������ֵ�ӽ������Dz���GW�㷨����һ���ľ����ԣ����㸴�Ӻ�ʱ�������ڴ˲�����GW�㷨������ʹ��Castep���������SnO2���ӽṹ�ķ���û��Ӱ�졣�����ܹ��ó����ӱ�Ϊ16.67%ʱ�õ��Ĵ�϶ֵΪ0.1301 eV����϶ֵ��δ����ʱ�������Խ��ͣ����Ҳ��Ӻ�ĵ����ͼ۴���Ŀ�������࣬��ø����ܼ����ܴ����ƽ���������˺ͼ۴����˻����ཻ��˵�����Ӻ�ʹ������������ϵ��ʾ�����ԡ�
ͼ5��6�ֱ������SnO2�Ͳ��ӱ�Ϊ16.67%ʱ��̬�ܶ�ͼ����̬�ܶ�ͼ����ͼ5���Կ��������ڷ����ܼ�����ߵ����ܼ���-15��-20 eV���۴��˵ķ�ֵ��Ҫ��O 2s̬�������ٲ���Sn 5s�Լ�Sn 5p̬�Ĺ��ף��߲��ӱ�ʱҲһ����ֻ�������䴦�����ܼ����Է����洦��Ӱ�켰С���ʿɺ��ԡ��ڷ�����8.3 eV��Χ�ڵļ۴��������3����ֵ��������߷���Ҫ����O 2p����Ĺ��ף���������ֵ��O 2p������ף�Ҳ��Sn 5s��Sn 5p������ף�����ռ�������ԱȲ�������£�O���������Ҫ����δ��������£���Sn���������Ҫ����δ��������£������������ܼ����ֶ���La 5d����Լ�O 2s������ף�ʹ����̬�ܶ�ͼ�ֶ���������ֵ���ڵ������֣�La 5p������뵼����Sn 5s��5p�����������δ����ʱ����С�����ҵ�������ܶ��ƶ��������Լ�����ͬʱ�ܹ�����С���ּ۴����뵼��������P�͵������ʣ�����Ѩ���硣�����Ϸ����ܹ������֪��Sn��O����̬���ף��ܹ��ó�SnO2�Ǿ���һ�������Ե����Ӽ�����[34-35]�����շ����õ�����ʹ�õ�������ܶ��ƶ����������ȱ�С����������ǿ�Ľ��ۣ��ﵽ�˲��Ӹ���SnO2�����Ե�Ŀ�ģ���������֤����ϡ��Ԫ��La�����ܹ�����AgSnO2��ͷ���ϵĵ����ԡ�
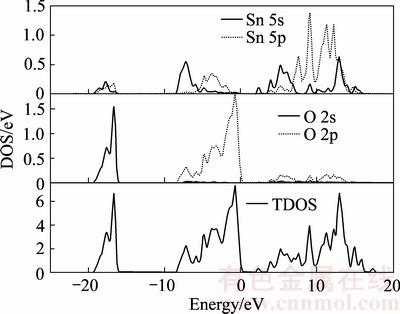
ͼ5 SnO2��̬�ܶ�ͼ
Fig. 5 Density of states(DOS) of SnO2

ͼ6 ���ӱ�Ϊ16.67%ʱSnO2̬�ܶ�ͼ
Fig. 6 Density of states(DOS) of 16.67% doped SnO2
��ģ�����õ��IJ�ͬ���ӱ���SnO2���ܴ�ͼ���ܹ��ó���������ȣ����5���С����Ӻ�ֱ�ӽ�������������С����϶����ֵ���Ų��ӱ������½����ֳ��ȼ�С������Ĺ��ɣ��ڲ��ӱ�Ϊ16.67%ʱ������������С��˵���������ɼ۴���������������������С����������ѡ�
��5 ��϶����
Table 5 Band gap parameters

��ģ�����õ���ͬ���ӱ���SnO2����̬�ܶ�ͼ����ͼ7��ʾ����ͼ7�п��Կ������Ų��ӱȵ����ӣ���̬�ܶ�ͼ�ĵ�������������ܼ������ƶ�����չ�������������ڵ������־���������ǿ�������ͬʱ�����ܼ��������������������Ӻ��С�������ܼ��������������������˲��ϵĵ����ԣ���̬�ܶȵĶԱ�ͼ�ܹ��ó��ڲ��ӱ�Ϊ16.67%ʱ��������ܼ������ƶ����������������С����������ѡ�

ͼ7 ��̬�ܶȶԱ�ͼ
Fig. 7 Comparison of total density of states(DOS)
2.4 La���ӵ�AgSnO2��ͷ������SnO2��������Ũ��
���ڴ�ͷ���϶��ԣ���������Խ��Ҫ���䵼���ԣ�������Ĺؼ���������Ũ�ȣ��������渽���ĵ���������������Ϊһ�ְ뵼����ϣ��䱾���������ǵ��ӺͿ�Ѩ���뵼�����ͨ�������ܹ��ı�����ӡ���Ѩ�������ܹ�����������Ũ�ȡ�ͨ��La���ӵĴ�ͷ������SnO2��̬�ܶ�ͼ�ܹ����������ܼ�����۴�����ϵ����P�͵������ʣ���ͨ����Ѩ���硣��ϵ�ķ����渽����������Ũ��Ϊ
 (1)
(1)
ʽ�У�VΪ�����������gc(E)Ϊ����������̬�ܶȣ������˷����浽5 eV��Χ�ڵ�������Ũ�ȣ����6��ʾ��
��6 ������Ũ��
Table 6 Carrier concentration

δ���ӵ�SnO2��������Ũ��Ϊ2.62��1018 cm-3(�������[36])���ɱ�6�ܹ��ó�����ʹ��������Ũ���������Ų��ӱȵļ�С��������Ũ�ȳ��ֳ���������С�Ĺ��ɣ��ڲ��ӱ�Ϊ16.67%ʱ������Ũ������ٴ�˵�����ӱ�Ϊ16.67%ʱ���ϵĵ���������ѡ�
3 ����
1) ���õ�һ��ԭ���Բ�ͬ������ϡ��La����SnO2���ϵľ���ṹ����ɲ��ӡ��ܴ��ṹ��̬�ܶȵȽ��м������������������������La���ӶԱ����뵼��SnO2���ʵĸı䡣
2) ��ͬ������La����SnO2������ֱ�Ӵ�϶�뵼����ϡ����Ӻ�����������La3+���Ӱ뾶�ϴ��йء�ͨ��Mulliken���ӷ���������ܶ�ͼ�ܹ��ó�����ʹ�ò������ֳ�һ���Ĺ����ԣ�����La��5d������뵼����ʹ�õ���������ܶ˷����ƶ����������ȱ�С�������Ӻ����������ֱ�Ӵ�϶�뵼����ϡ�
3) ͨ��������ͬ������La����SnO2�Ĵ�϶���Ⱥ�������Ũ�ȵó����ڲ��ӱ�Ϊ16.67%ʱ����������С�����������̶������ԾǨ��Ŀ��࣬��ϵ����P�͵������ʣ���Ҫͨ����Ѩ���磬˵���ڴ˲��������¶�SnO2�ĵ����Ը�����ѡ�
4) ͨ������SnO2�����ʣ����յó���AgSnO2��ͷ�����У������Ӽ��ı���ռSnO2��16.67%ʱ�����ϵ�������ѡ�
REFERENCES
[1] WOLF S A, AWSCHALOM D D, BUHRMAN R A, DAUGHTON J M,  S VON, ROUKES M L, CHTCHELKANVA A Y, TREGER D M. Spintronics: A spin-based electronics vision for the future[J]. Science, 2001, 294: 1488-1495.
S VON, ROUKES M L, CHTCHELKANVA A Y, TREGER D M. Spintronics: A spin-based electronics vision for the future[J]. Science, 2001, 294: 1488-1495.
[2] BAIBICH M N, BROTO J M, FERT A, NGUYEN V D F, PETROFF F, ETIENNE P, CREUZET G, CHAZELAS F A. Giant Magnetoresistance of (001) Fe/(001)Cr magnetic super lattices[J]. Physical Review Letters, 1998, 61: 2472-2475.
[3] �� ��, ���༪, �Ų���. N����SnO2���Ϲ�����ʵĵ�һ��ԭ���о�[J]. ����ѧ��, 2010, 10: 7285-7290.
YU Feng, WANG Pei-ji, ZHANG Chang-wen. First-principles study of optical and electronic properties of N-doped SnO2[J]. Acta Physica Sinica, 2010, 10: 7285-7290.
[4] �� ��, ���༪, �Ų���. Al����SnO2���ϵ��ӽṹ��ѧ����[J]. ����ѧ��, 2011(2): 210-216.
YU Feng, WANG Pei-ji, Zhang Chang-wen. Electronic structure and optical properties of Al-doped SnO2[J]. Acta Physica Sinica, 2011(2): 210-216.
[5] �� ��, �� ��, �Ŵ���. Cu��SnO2�Ĺ�������о�[J]. ������ͨ��, 2014, 12: 3103-3107.
MIAO Feng, HUANG Yi, ZHANG Chuan-wu. Research for photoelectric property of SnO2 doped Cu[J]. Bulletin of the Chinese Ceramic Society, 2014, 12: 3103-3107.
[6] �� ��, �� ��, ��ٻٻ, �� ��, �� ��, �� ��. Ru����Zr����Ԫ������ĵ�һ��ԭ���о�[J]. �й���ɫ����ѧ��, 2014, 24(5): 1327-1332.
WANG Xin, WANG Xin, YIN Qian-qian, WU Bo, LIN Wei, TANG Dian. First principles study of Zr-based binary oxide doped with Ru[J]. The Chinese Journal of Nonferrous Metals, 2014, 24(5): 1327-1332.
[7] WANG Li, FANG Li-hong, GONG Jian-hong. First-principles study of TiC(110) surface[J]. Transactions of Nonferrous Metals Society of China, 2012, 22(1): 170-174.
[8] YAN Hai-yan, WEI Qun, CHANG Shao-mei, GUO Ping. A first-principle calculation of structural, mechanical and electronic properties of titanium borides[J]. Transactions of Nonferrous Metals Society of China, 2011, 21(7): 1627-1633.
[9] �� ��, ���༪, �Ų���. ��һ��ԭ���������SnO2���ӽṹ��ѧ����[J]. ���ϴ�ѧѧ��(��Ȼ��ѧ��), 2009(4): 414-417.
YU Feng, WANG Pei-ji, ZHANG Chang-wen. First-principle study of optical and electronic properties of SnO2 [J]. Journal of University of Ji Nan (Sci & Tech), 2009(4): 414-417.
[10] ��ѧ��, ����Ƽ, ����, ����÷, �����. Ru����SnO2�뵼�������ĵ��ӽṹ�о�[J]. ����ѧѧ��, 2013, 12: 2514-2520.
XIE Xue-jia, ZHONG Li-ping, LIANG Zhen-hai, FAN Cai-mei, HAN Pei-de. Electronic structures of Ru-doped SnO2 semiconductor solid solutions[J]. Chinese Journal of Inorganic Chemistry, 2013, 12: 2514-2520.
[11] �� ÷, �ư�Ӣ. Sb��SnO2�Ĺ�������о�[J]. ���������ѧѧ��(��Ȼ��ѧ��), 2014(5: 768-771.
LONG Mei, YAN An-ying. Research on the photoelectric property of SnO2 doped Sb [J]. Journal of Southwest University for Nationalities (Sci and Tech), 2014(5): 768-771.
[12] ������, �͵´�, ���庭, ���. Ce����SnO2���Ͻṹ��ѧ���ʵ��о�[J]. ���, 2014(1): 25-28.
SHAN Lin-ting, BA De-chun, LIN Yi-han, LI Jian-chang. Structure and optical properties of Ce doped SnO2 [J]. Vacuum, 2014(1): 25-28.
[13] �ֽ�Ǭ, ��ѧ��, ����, ��С��, ����÷, �����. Ti����SnO2�뵼�������ĵ�һ��ԭ���о�[J]. �ߵ�ѧУ��ѧѧ��, 2012(5): 1050-1056.
JIA Jin-qian, XIE Xue-jia, LIANG Zhen-hai, ZHANG Xiao-chao, FAN Cai-mei, HAN Pei-de. First-principle study of Ti-doped SnO2 semiconductor solid solutions[J]. Chemical Journal of Chinese Universities, 2012(5): 1050-1056.
[14] �� ��, ֣ ��, �ο���, ������, ������, ���岨. ���ɽ�������SnO2�ĵ��ӽṹ�����[J]. ������ѧѧ��, 2010, 26(3): 763-768.
YU Li, ZHENG Guang, HE Kai-hua, ZENG Zhong-liang, CHEN Qi-li, WANG Qing-bo. Electronic structure and magnetism of transition metal doped SnO2[J]. Acta Physico- Chimica Sinica, 2010, 26(3): 763-768.
[15] �� ��, ������, ���༪. ���ɽ�������SnO2�������ѧ��ѧ���ʵ��о�[J]. ���ܲ���, 2014(3): 3070-3074.
WANG Ji, FENG Xian-yang, WANG Pei-ji. Study on magnetics and optical properties of transitionmetal doped SnO2 superlattice[J]. Functional Materials, 2014(3): 3070-3074.
[16] THANGARAJU B. Structure and electrical studies on highly consducting spray deposited fluorine and antimony doped SnO2 thin films from SnCl2 precursor[J]. Thin Solid Films, 2002, 402(1/2): 71-78.
[17] HAZEN R M, FINGER L W. Bulk moduli and high-pressure crystal structure of rutile-type compound[J]. Journal of Physics and Chemistry of Solids, 1981, 42(3): 143-151.
[18] BOLZAN A A, FONG C, KENNEDY B J. Structural studies of rutile-type metal dioxides[J]. Acta Crystallographica, 1997, 53(3): 373-380.
[19] �� ��, �� ƻ, �� ΰ, �ᄚ֥, ������. N-Al������TiO2���ӽṹ����ѧ���ʵ������о�[J]. �й���ɫ����ѧ��, 2015, 25(4): 1018-1024.
JIN Tao, ZHANG Ping, KAN Wei, TIAN Jing-zhi, DENG Qi-gang. Theory studies on electronic structure and optical properties of N-Al co-doped anatase TiO2[J]. The Chinese Journal of Nonferrous Metals, 2015, 25(4): 1018-1024.
[20] �����, ֣����, �� ��. Fe-S���������ѿ���TiO2�ĵ�һ��ԭ���о�[J]. �й���ɫ����ѧ��, 2013, 23(3): 852-858.
WU Guo-hao, ZHENG Shu-kai, LIU Lei. First-principles study on Fe-S co-doped anatase TiO2[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(3): 852-858.
[21] ��־��, �����, ������. �����ܶȷ������۵ĵ�һ��ԭ�����Ʒ�[J]. ������ѧ, 2005, 23(1): 1-4.
XIONG Zhi-hua, SUN Zhen-hui, LEI Min-sheng. First-principles with pseudopotentials method based on the density functional theory[J]. Jiangxi Scince, 2005, 23(1): 1-4.
[22] LIU Shu-bin. Conceptual density functional theory and some recent developments[J]. Acta Physico-Chimica Sinica, 2009, 25(3): 590-600.
[23] ������, �� ΰ, �����. �ܶȷ������ۼ�����ֵ�����½�չ[J]. ��ѧ��չ, 2005, 17(2): 192-202.
LI Zhen-yu, HE Wei, YANG Jin-long. Recent progress in density functional theory and its numerical methods[J]. Progress in Chemistry, 2005, 17(2): 192-202.
[24] ����, �� �, ��ǿ��. �ܶȷ������۵Ļ������㷽���о���չ[J]. �ɶ���֯�ߵ�ר��ѧУѧ��, 2015, 32(2): 39-43.
TANG Nan-nan, ZHANG Shu, LI Qiang-lin. Basic algorithms research progress of density functional theory[J]. Journal of Chengdu Textile College, 2015, 32(2): 39-43.
[25] ZHU Zhi-gang, RAMESH C D, ARUNABHIRAM C, RIADH S, HIDEYUKJ T, MICHIHISA K, NOZOMU H, AKIRA E, HIROMITSU T, CARLOS ADC, MOMOJ1 K, AKIRA M. Enhanced gas-sensing behavior of Ru-doped Sn02 surface: Aperiodic density functional approach[J]. Journal of Physics and Chemistry of Solids, 2009, 70: 1248-1255.
[26] MURAKAMI Y, ITO M, KAJI H, TAKASU Y. Surface characterization of ruthenium-tin oxide electrodes[J]. Applied Surface Science, 1997, 121: 314-318.
[27] ������, ½���, �� ��, ������. ��-ϡ�������ﴥͷ���ϵ�����[J]. ϡ�н��������빤��, 2001(3): 205-207.
WANG Jing-qin, LU Jian-guo, WEN Ming, WANG Bao-zhu. Study on the behavior of silver-rare earth oxide contact material[J]. Rare Metal Materials and Engineering, 2001(3): 205-207.
[28] ������, ������, �Ծ�Ӣ. Ag/SnO2-La2O3-Bi2O3��ͷ���ϵ��о�[J]. ϡ�н��������빤��, 2005, 10: 158-160.
WANG Hai-tao, WANG Jing-qin, ZHAO Jing-ying. Study on the Ag/SnO2-La2O3-Bi2O3 contact material[J]. Rare Metal Materials and Engineering, 2005, 10: 158-160.
[29] �� ��, ���༪, �Ų���, ������, �� ��, �Ź���. ������SnO2��Cr�ĵ��ӽṹ��ѧ���ʵ��о�[J]. ����ѧ��, 2011, 60(9): 227-232.
JIANG Lei, WANG Pei-Ji, ZHANG Chang-wen, FENG Xian-yang, LU Yao, ZHANG Guo-lian. Electronic structure and optical properties of Cr doped SnO2 superlattice[J]. Acta Physica Sinica, 2011, 60(9): 227-232.
[30] ������, Ҧ����, ����. SnO2���ӽṹ�����ѧ���Եĵ�һ��ԭ������[J]. ���ϿƼ�ѧԺѧ��(��Ȼ��ѧ��), 2011, 39(1): 78-82.
LIU Ya-ming, YAO Shu-wen, LIU Yu-bo. First-principles study of the electronic and optical properties of SnO2[J]. Journal of Henan Institute of Science and Technology, 2011, 39(1): 78-82.
[31] �� ��, ���༪, �Ų���, ������, �� ��, �Ź���. ��һ��ԭ���о�Fe����SnO2���ϵĹ������[J]. ����ѧ��, 2011, 11: 219-226.
LU Yao, WANNG Pei-ji, ZHANG Chang-wen, FENG Xian-yang, JIANG Lei, ZHANG Guo-lian. First-principles calculation on electronic structure and optical properties of iron-doped SnO2[J]. Acta Physica Sinica, 2011, 11: 219-226.
[32] DOLBEC R, KHAKANI MAE, SERVENTIA A M, TRUDEAU M, SAINT-JACQUES R G. Microstructure and physical properties of nanostructured tin oxide thin films grown by means of Pulsed laser deposition[J]. Thin Solid Films, 2002, 419: 230-236.
[33] BERGER J A, REINING L, SOTTILE F. Efficient GW calculations for SnO2, ZnO, and rubrene: The effective-energy technique[J]. Physical Review B: Condensed Matter, 2012, 85(8): 3711-3711.
[34] BARBARAT PH, MATAR S F. First-principle investigations of the electronic, optical and chemical bonding properties of SnO2[J]. Journal of Materials Chemistry, 1997, 7: 2547-2550.
[35] SENSATO F R, FILHO O T, LONGO E, SAMBRANO J R, ANDRES J. Theoretical analysis of the energy levels induced by oxygen vacancies and the doping process (Co, Cu and Zn) on SnO2 (110) surface models[J]. Journal of Molecular Structure Theochem, 2001, 541(1): 69-79.
[36] ��ѩ��, �˷���, ������, �� ��, �� ��, �� ��. Ru����Sn��������缫�ĵ�һ��ԭ������[J]. �й���ɫ����ѧ��, 2014, 24(5): 1333-1338.
LIU Xue-hua, DENG Fen-yong, WENG Wei-xiang, WANG Xin, LIN Wei, TANG Dian. First-principles calculation of Ru-doping Sn-based oxide electrode[J]. The Chinese Journal of Nonferrous Metals, 2014, 24(5): 1333-1338.
First-principles analysis of conductivity of La-doped AgSnO2 contact material
ZHAO Cai-tian, WANG Jing-qin, CAI Ya-nan, ZHOU Lu-lu, WU Qian
(Province-Ministry Joint Key Laboratory of Electromagnetic Field and Electrical Apparatus Reliability Hebei University of Technology, Tianjin 300130, China)
Abstract: AgSnO2 is an ideal substitute material for AgCdO electrical contact material. However, due to the near insulation of SnO2, the contact resistance of this material increases, so improving the electrical conductivity of SnO2 is an important problem to be solved. By using the first-principle method based on the density functional theory, different proportions (50%, 25%, 16.67%, 12.5%, 8.34%) of La doped SnO2 were established respectively, then lattice constant, Mulliken population, band structure and density of states were calculated. The results show that La doped SnO2 is also direct band gap semiconductor material, the band gap becomes smaller, the carrier concentration becomes larger, and the conductivity is enhanced. The doping ratio is 16.67% when the conductivity is the best.
Key words: La-doped SnO2; first-principle; electronic structure; electrical property
Foundation item: Project(E2016202106) supported by the Natural Science Foundation of Hebei Province, China; Project(ZD2016078) supported the Science and Technology Research of the Higher Education Institutions of Hebei Province, China
Received date: 2016-09-12; Accepted date: 2017-03-15
Corresponding author: WANG Jing-qin; Tel: +86-22-60204354; E-mail: jqwang@hebut.edu.cn
(�༭ ��ѧ��)
������Ŀ���ӱ�ʡ��Ȼ��ѧ������Ŀ(E2016202106)���ӱ�ʡ�ߵ�ѧУ��ѧ�����о���Ŀ(ZD2016078)
�ո����ڣ�2016-09-12�������ڣ�2017-03-15
ͨ�����ߣ������ۣ����ڣ���ʿ���绰��022-60204354��E-mail: jqwang@hebut.edu.cn

