从氯化镍溶液中电沉积α-Ni(OH)2
来源期刊:中国有色金属学报(英文版)2020年第10期
论文作者:张俊杰 张廷安 冯森
文章页码:2802 - 2811
关键词:α-Ni(OH)2;NiCl2;电沉积;初始pH;电流密度;电沉积温度;电沉积时间
Key words:α-Ni(OH)2; NiCl2; electrodeposition; electrolyte initial pH; current density; electrodeposition temperature; electrodeposition time
摘 要:采用电沉积方法从NiCl2溶液中合成α-Ni(OH)2。为了系统地研究实验条件的影响,选用一系列初始NiCl2溶液pH值、电流密度、电沉积温度和电沉积时间。循环伏安结果表明,存在副反应: Ni2++2e→Ni。从X射线衍射光谱分析、傅里叶红外光谱分析以及产物的颜色可知,当电解液初始pH为2~5.86、电流密度为10~25 mA/cm2、 电沉积温度为25~35 °C以及电沉积时间为1.0~3.0 h时,电沉积产物为纯净的α-Ni(OH)2。当沉积温度升高至45 °C时,获得的产物为α-Ni(OH)2和金属Ni的混合物;当电流密度超过30 mA/cm2时,获得产物呈β-Ni(OH)2的特征;当电流密度低至5 mA/cm2时,产物中有少量的金属Ni。电解液的pH随溶液的初始pH和电流密度的增加略微增加;产物的沉积质量随初始pH降低略微减小,随着电流密度的增加近乎线性增加。沉积质量-沉积时间曲线的斜率在起初的2.0 h内保持恒定,随后降低。
Abstract: α-Ni(OH)2 was synthesized from a NiCl2 solution by electrodeposition method. In order to conduct a systematic study on the effects of experimental parameters, a series of electrolyte initial pH values, current densities, electrodeposition temperatures, and electrodeposition time were used. Cyclic voltammetry results demonstrated a side reaction of Ni2++2e→Ni. The X-ray diffraction analysis, Fourier-transform infrared spectrum, and the color of the product showed that pure α-Ni(OH)2 could be obtained in the initial pH value range of 2-5.86, current density range of 10-25 mA/cm2, electrodeposition temperature range of 25-35 °C, and electrodeposition time range of 1.0-3.0 h. When electrodeposition temperature increased to 45 °C, a mixture of α-Ni(OH)2 and metallic Ni was obtained. A current density higher than 30 mA/cm2 resulted in the sample with features of β-Ni(OH)2. A small amount of metallic Ni existed in the as-prepared sample when current density decreased to 5 mA/cm2. A slight increase of electrolyte pH was observed with increasing initial solution pH and current density. Electrodeposition mass revealed a slight decrease with initial pH decreasing and showed an almost linear increase with current density increasing. The slope of the curve for electrodeposition mass versus electrodeposition time remained stable in the first 2.0 h and then decreased.
Trans. Nonferrous Met. Soc. China 30(2020) 2802-2811
Jun-jie ZHANG1,2, Ting-an ZHANG1,2, Sen FENG
1. School of Metallurgy, Northeastern University, Shenyang 110819, China;
2. Key Laboratory of Ecological Metallurgy of Multi-metal Intergrown Ores of Ministry of Education, Northeastern University, Shenyang 110819, China
Received 10 January 2020; accepted 7 August 2020
Abstract: α-Ni(OH)2 was synthesized from a NiCl2 solution by electrodeposition method. In order to conduct a systematic study on the effects of experimental parameters, a series of electrolyte initial pH values, current densities, electrodeposition temperatures, and electrodeposition time were used. Cyclic voltammetry results demonstrated a side reaction of Ni2++2e→Ni. The X-ray diffraction analysis, Fourier-transform infrared spectrum, and the color of the product showed that pure α-Ni(OH)2 could be obtained in the initial pH value range of 2-5.86, current density range of 10-25 mA/cm2, electrodeposition temperature range of 25-35 °C, and electrodeposition time range of 1.0-3.0 h. When electrodeposition temperature increased to 45 °C, a mixture of α-Ni(OH)2 and metallic Ni was obtained. A current density higher than 30 mA/cm2 resulted in the sample with features of β-Ni(OH)2. A small amount of metallic Ni existed in the as-prepared sample when current density decreased to 5 mA/cm2. A slight increase of electrolyte pH was observed with increasing initial solution pH and current density. Electrodeposition mass revealed a slight decrease with initial pH decreasing and showed an almost linear increase with current density increasing. The slope of the curve for electrodeposition mass versus electrodeposition time remained stable in the first 2.0 h and then decreased.
Key words: α-Ni(OH)2; NiCl2; electrodeposition; electrolyte initial pH; current density; electrodeposition temperature; electrodeposition time
1 Introduction
Nickel hydroxide is widely used as the active material for nickel-metal hydride (Ni-MH) rechargeable batteries due to its excellent electro- chemical performances [1-4]. It exists in two different crystallographic forms of α-Ni(OH)2 and β-Ni(OH)2 [5,6]. During the charging process, each Ni atom in α-Ni(OH)2 can exchange 1.69 electrons, resulting in a theoretical specific capacity of 482 mA・h/g [7,8], whereas each Ni atom in β-Ni(OH)2 can exchange 0.99 electron, leading to a theoretical capacity of 289 mA・h/g [9]. Therefore, by using α-Ni(OH)2 as the active material for Ni-MH batteries, a higher specific capacity can be achieved [10,11]. As portable energy storage devices are already a part of our daily life, and this trend will certainly increase, it is imperative to study the synthesis process of α-Ni(OH)2 to better meet this demand.
Numerous methods have been adopted to synthesize α-Ni(OH)2. Due to its low cost and short process time, electrodeposition is often used to prepare α-Ni(OH)2 [12-14]. Most commonly, Ni(NO3)2 is used as the raw material to prepare α-Ni(OH)2 via this method [15,16]. STREINZ et al [17,18] quantified the mass of nickel hydroxide as functions of electrolytic conditions by an electrochemical quartz crystal nanobalance (EQCN). JAYASHREE and KAMATH [19,20] studied the factors governing the electrodeposition process of α-Ni(OH)2 from Ni(NO3)2 solution and put forward the relevant electrodeposition mechanism. As we know, α-Ni(OH)2 is unstable in a strong alkaline solution and is progressively transformed into β-Ni(OH)2 [4,5]. JAYASHREE and KAMATH [21] investigated the effects of Al and Zn dissolved in the lattice of the electrodeposited α-Ni(OH)2 and found that α-Ni(OH)2 could be stabilized by Al and Zn. PAN et al [22] prepared Al-stabilized α-Ni(OH)2 from Ni(NO3)2 and Al(NO3)3 solutions by electrode- position method. GUALANDI et al [23] prepared multi-metals-stabilized α-Ni(OH)2 from its nitrate salts via electrodeposition.
SHANGGUAN et al [24] prepared α-Ni(OH)2 from various nickel salt solutions by a two-step drying method and reported that α-Ni(OH)2 prepared from NiCl2 solution exhibited better performance than that prepared from Ni(NO3)2 solution. Considerable attention has been paid to the preparation of α-Ni(OH)2 from Ni(NO3)2 solution; however, very few studies have investigated the synthesis route of α-Ni(OH)2 from NiCl2 solution by electrodeposition method. NiCl2 may be a more suitable raw material for synthesizing α-Ni(OH)2 by electrodeposition. YAO et al [25] synthesized α-Ni(OH)2・0.75H2O from a pure water dilute solution of NiCl2 (7-10 mmol/L) by electrodeposition method in a conventional three-electrode system and investigated the effects of electrolyte concentration and electrodeposition time. However, for industrial production, the reliability of this experiment needs to be considered. Besides the electrolyte concentration and reaction time, other conditions, electrolyte initial pH, current density, electrodeposition temperature, and electro- deposition time, should be considered.
In the present study, a systematic investigation of the experimental parameters effects on α-Ni(OH)2 electrodeposition was carried out.
2 Experimental
2.1 Preparation of materials
The electrolyte consisted of NiCl2・6H2O (analytical grade, ≥98%) as the solute and a mixture of ethanol and deionized water at the volume ratio of 1:1 (ethanol ≥99.7%) as the solvent [17,18,26]. The electrolytic cell was composed of a cathode chamber and two anode chambers. The anode chambers were located beside the cathode chamber. The chambers were separated with a cationic selectively permeable membrane. A stainless- steel sheet (35 mm × 90 mm × 1 mm) acted as the cathode and two iridium and ruthenium coated titanium sheets (17.5 mm × 90 mm × 1 mm) acted as the anode. In the present experiment, a series of the electrolyte initial pH values (2-5.86), current densities (5-30 mA/cm2), electrodeposition temperatures (25-45 °C), and electrodeposition time (1.0-3.0 h) were used. The concentration of the electrolyte was 0.2 mol/L (initial pH=5.86). In order to investigate the influences of the electrolyte initial pH, hydrochloric acid was used to acidize the electrolyte to pH values of 2, 3, 4, and 5. After electrodeposition, the synthesized powders in the cathode chamber were collected by filtration, washed with deionized water copiously, dried at 60 °C for 10 h, and weighed.
2.2 Measurements of electrodeposition system
A PHSJ-3F acidometer was used to record the pH of the cathodic electrolyte during the experimental process. An electrochemical workstation (Zennium-pro) was used for cyclic voltammetry (CV) measurements. CV measure- ments were carried out in a typical three-electrode system. Both the working electrode and counter electrode were made of the same material as that used in the electrodeposition process, and their diameters were 1 mm. An Ag/AgCl electrode was used as the reference electrode. The electrolyte used for CV measurements was composed of NiCl2 (1 g/L) and NaCl (1 g/L) dissolved in a solvent composed of ethanol and water at the volume ratio of 1:9. NaCl was used as the supporting electrolyte. All CV tests were run from negative to positive potentials at a scanning rate of 30 mV/s.
2.3 Characterization of materials
The phase of the prepared samples was confirmed via X-ray diffractometry (XRD) using a Bruker D8 X-ray diffractometer. Fourier-transform infrared spectroscopy (FT-IR) was performed using a Nicolet iS50 infrared spectrometer. The morphology of the products was observed using a scanning electron microscope (SEM; SU8010).
3 Results and discussion
3.1 CV curves

Fig. 1 CV curves of electrodeposition system with variations of initial pH in l g/L NiCl2 + 1 g/L NaCl solution (a), electrodeposition temperature in l g/L NiCl2 + 1 g/L NaCl solution (b), and solvents in 1 g/L NaCl solution (c)
The CV curves of the electrodeposition system presented in Fig. 1 reveal the occurrence of two cathodic reactions. The first corresponded to the reduction reaction of Ni2+ ions to metallic Ni at about -1.0 V (vs Ag/AgCl) (Eq. (1)) and the second corresponded to the hydrogen evolution reaction (Eq. (2)). In order to verify the hydrogen evolution reaction, other two CV curves of the working electrode were measured in the NiCl2-free electrolyte, as shown in Fig. 1(c). It is noticeable from Fig. 1(c) that the second reduction reaction was neither specific to the NiCl2 solution nor to the ethanol solution. It was even present in pure water solution of NaCl, which meant that it was a hydrogen evolution reaction. The hydogen evolution reaction occurred due to the cathodic reaction in the NiCl2 bath, and the first reduction reaction could be attributed to a side reaction of the electrodeposition process. Notably, in the acidic solution, the hydrogen evolution reaction should include Eqs. (2) and (3). As Eq. (2) dominated the hydrogen evolution reaction in the present work, it was used in the CV diagram to represent this reaction. OH- ions produced in Eq. (2) reacted with Ni2+ to form Ni(OH)2 (Eq. (4)), and the anodic reaction was the formation of chlorine gas (Eq. (5)).
Ni2++2e→Ni (1)
2H2O+2e→2OH-+H2 (2)
2H++2e→H2 (3)
Ni2++2OH-→Ni(OH)2 (4)
2Cl--2e→Cl2 (5)
The peak area of the side reaction in Fig. 1(a) increased with the decrease of the initial pH of the electrolyte; thus the number of Ni2+ ions taking part in the side reaction increased with the decreasing initial pH. The hydrogen evolution reaction showed similar trend with the side reaction. When the pH decreased, more H+ ions were generated in the electrolyte. OH- ions existed in a non-free state, thus more Ni2+ ions became free in these electrolytes. As the polarization became more negative, Ni2+ ions were reduced preferentially; hence, the reduction degree of Ni2+ ions increased with the decrease of the pH value. For the hydrogen evolution reaction, with the decreasing pH, the electrolyte produced more hydrogen as there were more H+ ions in the solution. Then, OH- ions became free by consuming H+ ions and reacted with Ni2+ to from Ni(OH)2 [27]. Figure 1(b) illustrates a favorable trend toward the side reaction with the increasing electrodeposition temperature, which is in accordance with the color of the prepared powders in Figs. 2(a1, a2). The color of the prepared samples changed from light green to dark green when the electrodeposition temperature increased from 35 to 45 °C. The dark green color was the result of a mixture of dark grey (the color of metallic Ni produced as a side product in this experiment) and light green (the color of Ni(OH)2). In addition to the initial pH of the electrolyte and the electrodeposition temperature, the current density also manifested a significant effect on the side reaction. The color of the product shifted from light green to celadon when the current density was reduced from 10 to 5 mA/cm2 (Fig. 2(b1, b2)). Figure 2(c1, c2) displays the color of the samples synthesized at initial pH values of 5.86 and 2. Based solely on the color changes of the samples, the effect of the initial pH on the side reaction cannot be distinguished.

Fig. 2 Photographs of samples obtained under different conditions
3.2 Phase constitution and structural disorder

Fig. 3 XRD patterns of samples synthesized in 0.2 mol/L NiCl2 solution under different conditions
It is well known that both the phase and average long-range structure of the sample can be exhibited by its XRD pattern [28]. Figure 3 displays the XRD patterns of the powder samples obtained under different electrodeposition conditions. The diffraction peaks at 2θ values of 22.7°, 33.4°, 38.7°, and 59.9° appeared from the (006), (101), (015), and (110) planes of pure hydrous nickel hydroxide (Ni(OH)2・0.75H2O, α-Ni(OH)2) (JCPDS No. 38- 0715). All of these powders obtained were indexed as pure α-Ni(OH)2 except for that obtained at 45 °C, which showed evident diffraction peaks of metallic nickel. Notably, the resultant powder obtained at a current density of 5 mA/cm2 was indexed as pure α-Ni(OH)2, which did not fit well with Fig. 2(b2). This discrepancy may result from the weak proportion of metallic nickel in the end product. From the aspect of the average long-range structure, it is evident that the initial pH of the electrolyte had no obvious effect on the diffraction peaks of the samples (Fig. 3(a)). This implied that both the average long-range structure and the degree of structural disorder remained stable with respect to the initial pH of the electrolyte. The diffraction peak intensities of the samples increased with increasing current density, electrodeposition temperature, and time (Figs. 3(b-d)). In particular, the diffraction peaks of the (006), (101), and (110) planes became narrow as the electrodeposition current density increased from 5 to 30 mA/cm2 (Fig. 3(b)) due to the supply of more OH- ions. In the presence of adequate OH- ions, the vacancies for OH- ions around a Ni atom tended to be fully occupied, facilitating the generation of a complete crystalline structure with no lattice defects.
3.3 FT-IR analysis
The diffraction peaks of α-Ni(OH)2 were not obvious due to its hydrotalcite-like structure (Fig. 3) [8,29]. FT-IR analysis provided the short- range coordination of the samples [28], and it complemented the XRD analysis. Both α-Ni(OH)2 and β-Ni(OH)2 possessed a layered structure stacked by Ni(OH)2 layers [30]; however, α-Ni(OH)2 existed in the OH- ion-deficient form [30,31]. In order to balance the excess positive charges, some anions (Cl- and CO32-) and water molecules were intercalated in the interspaces of Ni(OH)2 layers [24,32]. Figure 4(a) displays the FT-IR spectra of α-Ni(OH)2 and β-Ni(OH)2. In contrast to β-Ni(OH)2, α-Ni(OH)2 did not show the narrow absorption peak of non-H-bonded OH- groups at approximately 3640 cm-1; however, it showed a wide absorption peak of H-bonded OH- representing interlayer water molecules at approximately 3400 cm-1 [9,33]. In addition, compared to α-Ni(OH)2, the vibration peak of the in-plane Ni―O―H bond of β-Ni(OH)2 shifted from approximately 650 to 570 cm-1 [9,34]. For α-Ni(OH)2, the peaks at 1621 and 1361 cm-1 corresponded to the bending of water molecules and CO32- ions intercalated in the interlayer space (Figs. 4(b-d)) [4,35]. The very tiny absorption peak at 1062 cm-1 appeared from the vibration of the interlayered Cl- from the raw material. Figures 4(b) and (d) demonstrate that β-Ni(OH)2-free α-Ni(OH)2 was synthesized under varying initial pH (2-5.86) and electrodeposition time (1.0-3.0 h). Figure 4(c) shows the effect of current density on the FT-IR spectra of the products. A dimly-visible narrow peak at 3640 cm-1 representing β-Ni(OH)2 appeared at the current density of 30 mA/cm2, indicating that further increasing current density could lead to β-Ni(OH)2.

Fig. 4 FT-IR spectra of α-Ni(OH)2 and β-Ni(OH)2 (a), and samples obtained under different electrolyte initial pH values (b), current densities (c), and electrodeposition time (d)
3.4 Morphology of synthesized samples
The morphologies of the samples obtained under different electrodeposition conditions manifested no prominent difference (Fig. 5). They all consisted of large and small irregularly-shaped particles, similar to those obtained from a Ni(NO3)2 solution by the electrodeposition method [20,22].
3.5 Variation of electrolyte pH
The curves of pH versus time for the cathodic electrolyte at various initial pH values and current densities are illustrated in Fig. 6. The solid and dashed lines in Fig. 6 represent the measured (the metrical position is 2.5 cm from the cathode) and calculated results based on the solubility product constant (Ksp) of Ni(OH)2 (5.5×10-16), respectively. It is evident from Fig. 6 that the measured values of the pH were lower than the calculated value, which indicated that theoretically, no Ni(OH)2 was synthesized in the bulk solution of the cathodic chamber. In fact, Ni(OH)2 was deposited on the surface of the electrode and then was detached to the vicinity of the electrode (Fig. 6(a)). The metrical position of the pH value could be considered to be in the bulk solution because there was a distance of 2.5 cm between them. Figure 6(a) reveals that the pH of the electrolyte remained stable after 5 min of electrodeposition even when the initial pH value was 2. In comparison to the unacidified electrolyte (pH=5.86), more H+ ions were present in the electrolyte acidified by hydrochloric acid; thus this acidified electrolyte easily produced more H+ ions (Fig. 6(a)), thereby these H+ ions were quickly consumed, consequently, OH- ions became free in the electrolyte. The pH of the electrolyte began to increase until the generation and consumption rates of OH- became almost the same; therefore, the initial pH almost had no effect on the pH of the electrolyte after 5 min electrodeposition. Notably, the electrolytic energy consumed by H+ ions did not result in OH- ions, leading to a waste of energy. Figure 6(b) presents the relationship between the pH of the cathodic electrolyte and the current density. A slight increase in the pH was observed with the increasing current density, because OH- ions produced on the electrode surface were not consumed in time and were transported to the bulk solution.

Fig. 5 SEM images of samples obtained under different electrodeposition conditions
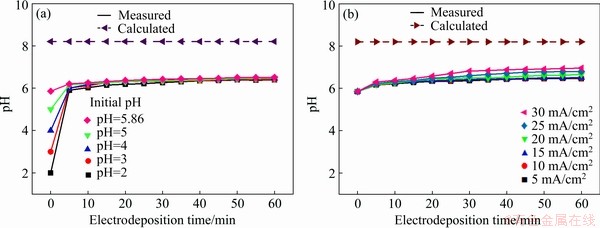
Fig. 6 Variation of pH of electrodeposition systems vs time at different initial pH (a) and current densities (b)
3.6 Deposition mass versus electrodeposition conditions

Fig. 7 Relationship between electrodeposition mass and electrodeposition conditions with varying electrolyte initial pH (a), current density (b), and electrodeposition time (c)
Figure 7 shows the relationship between the deposition mass and different electrodeposition conditions. The solid and dashed lines in Fig. 7 represent measured and calculated results based on Faraday’s law, respectively. It is noticeable that the collected mass of each separated sample decreased slightly with the decrease of initial pH (Fig. 7(a)) because the consumption of electrolytic energy by H+ ions used to acidify the solution. Figure 7(b) shows an almost linear relationship (R2=0.9993) between the electrodeposition mass and the current density (10-30 mA/cm2). The slope of curve for electrodeposition mass vs electrodeposition time (electrodeposition rate) remained nearly constant when the electrodeposition time was less than 2.0 h and decreased after that, as shown in Fig. 7(c), because the concentration of Ni2+ was decreased due to consumption. Particularly, some metrical points were found to be higher than the calculated ones (Fig. 7) because the actual molecular mass is higher than the applied one (106.19 for Ni(OH)2・0.75H2O) due to the presence of intercalated anions (CO32-, Cl-) (Fig. 4) [17,18,20].
4 Conclusions
(1) CV results demonstrated a side reaction of Ni2++ 2e→Ni.
(2) XRD results showed that pure α-Ni(OH)2 was obtained in the initial pH value range of 2-5.86, the current density range of 5-30 mA/cm2, the electrodeposition temperature range of 25-35 °C, and the electrodeposition time range of 1.0-3.0 h. When the electrodeposition temperature increased to 45 °C, a mixture of α-Ni(OH)2 and metallic Ni was obtained. FT-IR results showed that current density of 30 mA/cm2 resulted in samples with features of β-Ni(OH)2. The color of the product displayed that a small amount of metallic Ni existed in the sample when the current density decreased to 5 mA/cm2.
(3) The pH of the cathodic electrolyte displayed a slight increase with increasing electrolyte initial pH and current density.
(4) Electrodeposition mass showed no obvious influence by the initial pH of the electrolyte and increased almost linearly with the current density increasing. The slope of curve of electrodeposition mass versus electrodeposition time remained stable in the first 2.0 h and decreased after that.
References
[1] ASH B, KHETIC K, SUBBAIAH T, ANAND S, PARAMGURU R K. Physcio-chemical and electro-chemical properties of nickel hydroxide precipitated in the presence of metal additives [J]. Hydrometallurgy, 2006, 84: 250-255.
[2] LIU B, YUAN H T, ZHANG Y S, ZHOU Z X, SONG D Y. Cyclic voltammetric studies of stabilized α-nickel hydroxide electrode [J]. Journal of Power Sources, 1999, 79: 277-280.
[3] HU W K, GAO X P, NOREUS D, BURCHARDT T, NAKSTAD N K. Evaluation of nano-crystal sized α-nickel hydroxide as an electrode material for alkaline rechargeable cells [J]. Journal of Power Sources, 2006, 160(1): 704-710.
[4] LI J L, ASLAM M K, CHEN C G. One-pot hydrothermal synthesis of porous α-Ni(OH)2/C composites and its application in Ni/Zn alkaline rechargeable battery [J]. Journal of the Electrochemical Society, 2018, 165(5): A910-A917.
[5] YAO J H, LI Y W, LI Y X, ZHU Y X, WANG H B. Enhanced cycling performance of Al-substituted α-nickel hydroxide by coating with β-nickel hydroxide [J]. Journal of Power Sources, 2013, 224: 236-240.
[6] BAO Jie, ZHU Yan-juan, ZHUANG Yi-huan, XU Qing-sheng, ZHAO Ru-dong, LIU Yong-lin, ZHONG Hao-liang. Structure and electrochemical performance of Cu singly doped and Cu/Al co-doped nano-nickel hydroxide [J]. Transactions of Nonferrous Metals Society of China, 2013, 23: 445-450.
[7] CHENG F Y, CHEN J, SHEN P W. Y(OH)3-coated Ni(OH)2 tube as the positive-electrode materials of alkaline rechargeable batteries [J]. Journal of Power Sources, 2005, 150: 255-260.
[8] HUANG H L, GUO Y J, CHENG Y H. Ultrastable α phase nickel hydroxide as energy storage materials for alkaline secondary batteries [J]. Applied Surface Science, 2018, 435: 635-640.
[9] LI Y W, YAO J H, ZHU Y X, ZOU Z G, WANG H B. Synthesis and electrochemical performance of mixed phase α/β nickel hydroxide [J]. Journal of Power Sources, 2012, 203: 177-183.
[10] LI Y W, YAO J H, LIU C J, ZHAO W M, DENG W X, ZHANG S K. Effect of interlayer anions on the electrochemical performance of Al-substituted α-type nickel hydroxide electrodes [J]. International Journal of Hydrogen Energy, 2010, 35: 2539-2545.
[11] JAYASHREE R S, KAMATH P V. Nickel hydroxide electrodeposition from nickel nitrate solutions: Mechanistic studies [J]. Journal of Power Sources, 2001, 93: 273-278.
[12] DONG Q, YUAN H L, YAN Y B. Research progress in preparation methods of nickel hydroxide [J]. Hebei Chemical, 2004, 6: 28-31. (in Chinese)
[13] DIXIT M, KAMATH P V, KUMAR V G, MUNICHANDRAIAH N, SHUKLA A K. An electrochemically impregnated sintered-nickel electrode [J]. Journal of Power Sources, 1996, 63: 167-171.
[14] SASAKI Y, YAMSHITA T. Effect of electrolytic conditions on the deposition of nickel hydroxide [J]. Thin Solid Films, 1998, 334: 117-119.
[15] ASH B, MISHRA K G, SUBBAIAH T, PARAMGURU R K, MISHRA B K. Electrochemical studies on electrolytic preparation of battery grade nickel hydroxide―Effect of OH- to Ni2+ ratio [J]. Journal of Power Sources, 2015, 275: 55-63.
[16] WATANABE K, KIKUOKA T, KUMAGAI N. Physical and electrochemical characteristics of nickel hydroxide as a positive material for rechargeable alkaline batteries [J]. Journal of Applied Electrochemistry, 1995, 25: 219-226.
[17] STREINZ C C, MOTUPALLY S, WEIDNER J W. The effect of temperature and ethanol on the deposition of nickel hydroxide films [J]. Journal of the Electrochemical Society, 1995, 142: 4051-4056.
[18] STREINZ C C, HARTMAN A P, MOTUPALLY S, WEIDNER J W. The effect of current and nickel nitrate concentration on the deposition of nickel hydroxide [J]. Journal of the Electrochemical Society, 1995, 142: 1084-1089.
[19] JAYASHREE R S, KAMATH P V. Factors governing the electrochemical synthesis of α-nickel(II) hydroxide [J]. Journal of Applied Electrochemistry, 1999, 29: 449-454.
[20] JAYASHREE R S, KAMATH P V. Nickel hydroxide electrodeposition from nickel nitrate solutions: Mechanistic studies [J]. Journal of Power Sources, 2001, 93: 273-278.
[21] JAYASHREE R S, KAMATH P V. Suppression of the α→β-nickel hydroxide transformation in concentrated alkali: Role of dissolved cations [J]. Journal of Applied Electrochemistry, 2011, 31: 1315-1320.
[22] PAN T, WANG J M, ZHAO Y L, CHEN H, XIAO H M, ZHANG J Q. Al-stabilized α-nickel hydroxide prepared by electrochemical impregnation [J]. Materials Chemistry and Physics, 2003, 78: 711-718.
[23] GUALANDI I, MONTI M, SCAVETTA E, TONELLI D, PREVOT V, MOUSTY C. Electrodeposition of layered double hydroxides on platinum: Insights into the reactions sequence [J]. Electrochimica Acta, 2015, 152: 75-83.
[24] SHANGGUAN E B, LI J, GUO D, GUO L T, NIE M Z, CHANG Z R, YUAN X Z, WANG H J. A comparative study of structural and electrochemical properties of high-density aluminum substituted α-nickel hydroxide containing different interlayer anions [J]. Journal of Power Sources, 2015, 282: 158-168.
[25] YAO K L, ZHAI M H, NI Y H. α-Ni(OH)2・0.75H2O nanofilms on Ni foam from simple NiCl2 solution: Fast elelctrodeposition, formation mechanism and application as an efficient bifunctional electrocatalyst for overall water splitting in alkaline solution [J]. Electrochimica Acta, 2019, 301: 87-96.
[26] PAN T. Preparation of stabilized α-Ni(OH)2 by cathodic electrodeposition [D]. Hangzhou: Zhejiang University, 2003. (in Chinese)
[27] YAVUZ A, ERDOGAN P Y, OZDEMIR N, ZENGIN H, ZENGIN G, BEDIR M. Electrochemical synthesis of CoOOH-Co(OH)2 composite electrode on graphite current collector for supercapacitor applications [J]. Journal of Materials Science: Materials in Electronics, 2019, 30: 18413-18423.
[28] RAMESH T N, VISHNU KAMATH P. Synthesis of nickel hydroxide: Effect of precipitation conditions on phase selectivity and structural disorder [J]. Journal of Power Sources, 2006, 156: 655-661.
[29] MIYAATA S. Anion-exchange properties of hydrotalcite-like compounds [J]. Clays and Clay Minerals, 1983, 31: 305-311.
[30] LI H L, LIU S Q, HUANG C H, ZHOU Z, LI Y H, FANG D. Characterization and supercapacitor application of coin-like β-nickel hydroxide nanoplates [J]. Electrochimica Acta, 2011, 58: 89-94.
[31] ANDRATE T M, DANCZUK M, ANAISSI F J. Effect of precipitating agents on the structural, morphological, and colorimetric characteristics of nickel hydroxide particles [J]. Colloid and Interface Science Communications, 2018, 23: 6-13.
[32] HU M, YANG Z Y, LEI L X, SUN Y M. Structural transformation and its effects on the electrochemical performances of a layered double hydroxide [J]. Journal of Power Sources, 2011, 196: 1569-1577.
[33] PATNAIK P, BISWAL A, TRIPATHY B C, PRADHAN S, DASH B, SAKTHIVEL R, SUBBAIAH T. Synthesis and characterization of fibrous nickel hydroxide obtained from spent nickel catalyst [J]. Transactions of Nonferrous Metals Society of China, 2013, 23: 2977-2983.
[34] MIAO C C, ZHU Y J, HUANG L G, ZHAO T Q. The relationship between structural stability and electrochemical performance of multi-element doped alpha nickel hydroxide [J]. Journal of Power Sources, 2015, 274: 186-193.
[35] ZHAO Y L, WANG J M, CHEN H, PAN T, ZHANG J Q, CAO C N. Al-substituted α-nickel hydroxide prepared by homogeneous precipitation method with urea [J]. International Journal of Hydrogen Energy, 2004, 29: 889-896.
张俊杰1,2,张廷安1,2,冯 森1
1. 东北大学 冶金学院,沈阳 110819;
2. 东北大学 多金属共生矿生态冶金教育部重点实验室,沈阳 110819
摘 要:采用电沉积方法从NiCl2溶液中合成α-Ni(OH)2。为了系统地研究实验条件的影响,选用一系列初始NiCl2溶液pH值、电流密度、电沉积温度和电沉积时间。循环伏安结果表明,存在副反应: Ni2++2e→Ni。从X射线衍射光谱分析、傅里叶红外光谱分析以及产物的颜色可知,当电解液初始pH为2~5.86、电流密度为10~25 mA/cm2、 电沉积温度为25~35 °C以及电沉积时间为1.0~3.0 h时,电沉积产物为纯净的α-Ni(OH)2。当沉积温度升高至45 °C时,获得的产物为α-Ni(OH)2和金属Ni的混合物;当电流密度超过30 mA/cm2时,获得产物呈β-Ni(OH)2的特征;当电流密度低至5 mA/cm2时,产物中有少量的金属Ni。电解液的pH随溶液的初始pH和电流密度的增加略微增加;产物的沉积质量随初始pH降低略微减小,随着电流密度的增加近乎线性增加。沉积质量-沉积时间曲线的斜率在起初的2.0 h内保持恒定,随后降低。
关键词:α-Ni(OH)2;NiCl2;电沉积;初始pH;电流密度;电沉积温度;电沉积时间
(Edited by Wei-ping CHEN)
Foundation item: Project (U1710257) supported by the National Natural Science Foundation of China
Corresponding author: Ting-an ZHANG; Tel: +86-24-83681563; E-mail: zta2000@163.net
DOI: 10.1016/S1003-6326(20)65422-X

