DOI: 10.11817/j.ysxb.1004.0609.2021-40035
�Ͼ������������ѧ����Ӱ��ķ��Ӷ���ѧģ��
Ѧ����������ǿ����·������˼������ Ƽ
(�Ϸʹ�ҵ��ѧ ���Ͽ�ѧ�빤��ѧԺ���Ϸ� 230009)
ժ Ҫ��Ϊ���о��Ͼ����Ĵ�С����������ѧ���ܼ����λ�����Ӱ�죬���÷��Ӷ���ѧ�Բ�ͬ�Ͼ������Ͼ��ٽ����˵�������ģ�⡣ʹ�ý����б�����(CNA)��λ����������(DXA)����������������ٵı���ʧЧ���̺��ṹ�ݻ������˱����������Ӷ���ʾ�Ͼ�������������ѧ����Ӱ���ۻ���������������Ͼ��ٱ��ι����г��ֵ���䡢�Ͼ���ı����Լ�ȥ�Ͼ����������ı��Ͼ��������Ƶ���չ��ʽ������Ͼ���ı����������������Ͼ����ļ�С���Ͼ��ܶȵ����ӣ��ɱ��ε��Ͼ������࣬���������Ͼ��ٵĶ���Ӧ�����ӡ������Ͼ����д��������ϸߵ�����õ�������ԭ�ӽṹʹ�������и����׳��־���ȱ�ݣ�ȱ�ݻ��������غ������¿����γ����ƣ����¾������ʧЧ�����ؽ����������ٵ�����ǿ�ȡ����⣬�Ͼ���Ĵ������������˼��α���λ��������ͬʱ�谭��λ���Ļ����˶���λ�����Է�����˶����Ӷ��������Ա�
�ؼ��ʣ��٣������Ͼ��磻�Ͼ��ࣻλ�������Ӷ���ѧ
���±�ţ�1004-0609(2021)-08-2136-10���� ��ͼ����ţ�TG147���� ���ױ�־�룺A
���ĸ�ʽ��Ѧ����, ����ǿ, ��·��, ��. �Ͼ������������ѧ����Ӱ��ķ��Ӷ���ѧģ��[J]. �й���ɫ����ѧ��, 2021, 31(8): 2136-2145. DOI: 10.11817/j.ysxb.1004.0609.2021-40035
XUE Ke-min, ZHANG Yong-qiang, WANG Lu-sheng, et al. Molecular dynamics simulation of effect of twin boundaries spacing on mechanical properties of nano-tungsten[J]. The Chinese Journal of Nonferrous Metals, 2021, 31(8): 2136-2145. DOI: 10.11817/j.ysxb.1004.0609.2021-40035
��(W)���и��۵㡢���ܶȡ���Ӳ�ȡ���ģ������������ϵ������ĥ����ʴ�����õ���������ѧ�ͻ�е���ܣ����㷺Ӧ���ں��պ��졢����װ�����˹�ҵ���ڶ������ǹ����Ⱥ˾۱䷴Ӧ��(ITER)�������Ҫ��ѡ����[1-3]���������۵�ϸߣ���ҵ��ͨ�����÷�ĩұ��ķ��������Ʊ����ս����д��ڴ����Ŀ������ʣ����������Ա��������ϲ����Ӱ��������Ժ�������[4-6]��ͬʱ���־��ս��ٴ��������صĵ��´��Ժ��ٽᾧ���Եȴ����[7]��������Լ�˴��ٵļӹ���Ӧ�á����ô����Ա���(SPD)���������ڲ��ı��仯ѧ�ɷֵ����������ٻ����ϵ���ѧ����[8-9]�����Ŷ��ڲ��ô����Ա��ι������ı䴿�ٵ�����ʱ�����������ڱ��κ�������й۲쵽�Ͼ��Ĵ��ڡ�
�Ͼ�����Ϊһ������Ĵ�ǶȾ��磬�㷺�ش����ڸ��ֳ��õĽ������ϵ��С�BEAVEN��[10]����FIM-TEM(Field ion microscopy and transmission electron microscopy)���������״ι۲쵽�����ڽ������е��Ͼ��磬�����ط������Ͼ��紦���ڵ�ȱ�ݽṹ��MIKHAILOVSKIJ��[11]��WENSKY��[12]��KRASKO[13]�ֱ����÷��Ӿ�̬ģ�ⷽ���о��˾��������Ͼ����ԭ���Ų���ʽ��������Ħ��ࡰ�����Ρ��ṹ������ȫ�����������Ͼ����ԭ�ӹ��ͣ����Ͼ����渽���Ľ���ԭ������Ժϲ��γ����������ṹ��ԭ���档��ͳ�Ĺ���ǿ�����ڶ���ǿ�����α�ǿ���ȶ����������������ȱ�ݣ�ʹ������ǿ�����ӵ�ǿ�����ƣ�������ȱ�ݵ����ӣ�λ�������˶�������ѣ����ն�������������Ե��½�[14-15]���������Ͼ���ǿ����Ϊһ������ǿ�����������������⡣���о�����[17]��������������Ͼ����ܹ������ӽ���ǿ�ȵ�ͬʱ��������������˽��������ԡ�XU��[17]��WANG��[18]�о����֣���������Ͼ���(CTB)�����ظı��˽������ϵ���ѧ�͵�ѧ���ʣ�����CTB���������������غ�ʱ����ǿ�Ȳ�������Ͼ����(CTBs)�仯��������ѹ���غ�ʱ��������Ͼ�������Ƶ�����õ�������ǿ����CTBs�ļ�С������
���Ӷ���ѧ���о��ij߶��������ģ��ܹ�ֱ�۵�չ�־������ȱ�ݺ;���ṹ���ݱ���̣�������÷��Ӷ���ѧģ��ķ������о��Ͼ���������������ѧ���ܼ����λ�����Ӱ��������ơ���껷ɵ�[19]���÷��Ӷ���ѧ�����о����ݶ������Ͼ�ͭ�������غ��µı��μ����ݻ����̣�֤���Ͼ��Ĵ��ڿ������ͭ�����ԣ������Ͼ��ܶȵ����ӣ��Ͼ�ͭ��ǿ�Ⱥ�Ӳ��Ҳ��������ߡ����캵��[20]���÷��Ӷ���ѧ�����Բ�ͬ�Ͼ�������������е�������ģ�⣬���ֵ���ģ��������ǿ�ȶ����Ͼ�������Ӷ����ӣ��ҵ�����������ǿ����ߡ�
��Ȼ����ѧ�߶������Ͼ�����չ�����о����������Ͼ����о�������Ҫ���������������ṹ(FCC)DE1�����������ṹ(HCP)�Ľ���[21-22]���������������ṹ(BCC)�������Ͼ�ǿ�����Ƶ��о�����Խ��١�����BCC�������Ͼ��������������α��Ӱ�켰������û��ȷ���Ľ��ۡ��������÷��Ӷ���ѧ�����о������������ṹ�����Ͼ����ڵ��������غ������µı��μ�ʧЧ��Ӱ�죬���ڶ������Ͼ����������غ��µ��۱��λ����ṩ���۲ο���
1 ���Ӷ���ѧ��ģ����
1.1 �����Ͼ���ԭ��ģ��
Ϊ���о��Ͼ���������������������غ������µ���ѧ���ܺ��۱���Ӱ������������˲�ͬ�Ͼ���˵ķ��Ӷ���ѧģ�ͣ��˵ķ�Χ��1.09 nm��8.2 nm[17, 19-20]�������Ͼ����(Twin boundary spacing, TBS)Ϊ2.73 nm��ģ����ͼ1��ʾ�������ٵĵ����ṹΪ���������ṹ���������Ϊa=b=c=3.165  �������ºͦ�Ϊ90�㡣ģ�͵�����ߴ�ΪLa=19.4 nm��Lb=16.4 nm��Lc=13.4 nm��ģ���е�ԭ������Ϊ248200����������ľ�����x��y��z�ľ���ֱ�Ϊ
�������ºͦ�Ϊ90�㡣ģ�͵�����ߴ�ΪLa=19.4 nm��Lb=16.4 nm��Lc=13.4 nm��ģ���е�ԭ������Ϊ248200����������ľ�����x��y��z�ľ���ֱ�Ϊ ��[111]��
��[111]�� ��
��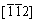 ��[111]��
��[111]��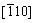 ��
��
1.2 �ƺ������߽���������

ͼ1 �Ͼ����Ϊ2.73 nm�ķ��Ӷ���ѧģ��
Fig. 1 Molecular dynamics model of twin boundary spacing ��=2.73 nm
������LAMMPS (Large-scale atom/scaler massively parallel simulation)[23-24]�������з��Ӷ���ѧģ�⡣LAMMPSģ�������������ƺ�����ѡ����Ϊ��Ҫ�����ڴ�����������ԣ�Ƕ����(Embedded atomic method��EAM)�ƺ������õ����[25-27]��EAM�Ʒ����а�ÿ��ԭ�ӵ�������Ƕ�����¼����еĸ��壬ͨ��һϵ�к��������ø������������ѧ��ϵ����ϵ����������Ϊ��Ƕ�ܺ����������֮��[28]������Ƕ��ԭ���ƵĶ��壬ϵͳ������(U)���Ա�ʾΪ
 (1)
(1)
ʽ�У��ұߵ�һ��Fi��Ƕ���ܣ��ڶ����Ƕ���� ����������ԭ�ӵĺ�������ڵ�i��ԭ�Ӵ������ĵ������ܶ�֮�ͣ������Ա�ʾΪ
����������ԭ�ӵĺ�������ڵ�i��ԭ�Ӵ������ĵ������ܶ�֮�ͣ������Ա�ʾΪ
 (2)
(2)
ʽ�У� �ǵ�j��ԭ�ӵĺ�������ڵ�i��ԭ�Ӵ��ĵ���ܶȡ�
�ǵ�j��ԭ�ӵĺ�������ڵ�i��ԭ�Ӵ��ĵ���ܶȡ�
��ģ��Ϊ������W-W֮���������ƣ�����ZHOU��[29]�Ľ���EAM�ƣ����ƺ����Ѿ��㷺Ӧ���������ٵ����졢ѹ������ѧ���ܵ��о�[1, 17, 25, 30]��
����Verlet-velocity����[31]����ԭ��������˶��켣��ţ�ٷ��̡�����ģ���Ϊ�����ֽ��У�������NVEϵ���¶�ģ�ͳ�ֳ�ԥʹ�ṹ�ﵽ������С���ͣ�ʱ�䲽��Ϊ0.001 ps����ԥʱ��Ϊ40 ps���ڳ�ԥ��ɺ�����ģ�������NVTϵ���½�������ģ�⣬������ϵ���¶�����Ϊ300 K�������б߽�����Ϊѭ�������Ա߽磬ÿ1000 �����ԭ�����ꡢ�¶ȡ����ܺͶ��ܵ���Ϣ��ͨ�����Է�������(Common neighbor analysis��CNA)��λ��������(Dislocation analysis��DXA)������ԭ���ڲ��ṹ�ı仯[32]��ģ��ͨ��LAMMPS��Դ������̽��м��㣬����OVITO�����������п��ӻ�����[33]��
2 ���������
2.1 �Ͼ���������ѧ���ܵ�Ӱ��
���������£��ٵ�����Ӧ��-Ӧ�������ܹ��ܺõط�Ӧ�����������ѧ���ܣ�ͨ����ģ�ͽ���������Ӷ���ѧģ����Եõ���ͬ�Ͼ��������ٺ͵����ٵ�Ӧ��-Ӧ�����ߺ͵���ģ�����ߡ�ͼ2��ʾΪ�����ٺͲ�ͬTBS�Ͼ����ڵ��������غ��µ���ѧ���ܡ�ͼ2(a)��ʾΪ300 K�²�ͬTBS�Ͼ��ٺ͵���������Y�᷽���������غɵ�Ӧ��-Ӧ�����ߣ�ͼ2(b)��ʾΪ�䵯��ģ�����ߡ���ͼ2������1) �����ٺ��Ͼ��ٵ�ʧЧ��ʽ���Ǵ��Զ��ѣ������ٵ�ʧЧӦ��Ϊ17%���Ͼ��ٵ�ʧЧӦ������TBS�ļ�С��������ԶԶС�ڵ����١�2) �Ͼ����Ц�=1.09 nmʱʧЧӦ������Ͼ��ٵ�ʧЧӦ����TBS���������С������TBS�ﵽ5.46 nmʱ�Ͼ��ٵ�ʧЧӦ��Ͳ������ӻ������ӵ��ٶȷdz�������3) �Ͼ��ٵ�����ǿ��Ϊ28 GPa��ҲԶԶС�ڵ����٣��Ͼ��ٵ�����ǿ����TBS�Ĵ�С�����ء�����XU��[17]�������Ͼ������о������ƵĽ������ͬ���������ǵ��о����Ͼ����Ϊ1 nm����ʱ���Ͼ��ٵ�����ǿ�ȸ����ҳ�����Ӧ��Ӳ�����̣����ֳ��������Ͼ��ٲ�һ���ı��ι��̡�4) �Ͼ��ٵĵ���ģ������TBS��������������������ڵ����٣����ˣ�3.28 nmʱ����ģ����TBS�������������ʽϿ졣����CHEN��[34]���о���֪�����ڲ����Ͼ��������û������Ͼ��������������������Ͼ���֮����ڴ�СΪ1/��2�������������������ı��Ͼ������̬�����ˣ�3.28 nmʱ����ģ����TBS���Ӷ����ӵ����ʼ�С���������˴ﵽ5.46 nm���Ͼ��ٵĵ���ģ���������Ͼ�������Ӷ����ӣ���ʱ�����Ͼ���֮�������ý�С��
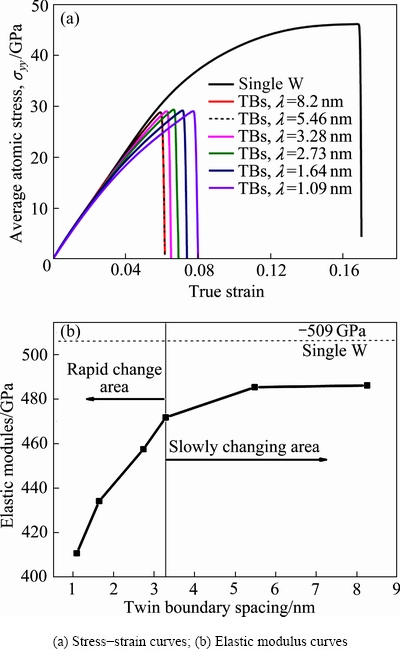
ͼ2 �����ٺͲ�ͬTBS�Ͼ����ڵ��������غ��µ�Ӧ��-Ӧ������
Fig. 2 Stress-strain curves of single crystal and twin tungsten with different TBS under uniaxial tension
2.2 ��ͬ�Ͼ����������������ʧЧ���̵ľ����۽ṹ
ͼ3��ʾΪ�����ٺͲ�ͬTBS���Ͼ������ܵ��������غ��µı���ʧЧ���̡�ͼ3(a)��ʾΪ�������������ʧЧ���̡��������ٵ�Ӧ��ﵽ16.9%ʱ���ھ����ڲ��IJ�ͬλ��ͬʱ��ʼ����һЩ��λȱ�ݡ���Ӧ��ﵽ16.95%ʱ����λȱ�ݶ�����[210]������չ��Ϊ���ƣ��������Ƶĸ�����������������������(HCP)������١�����Ӧ���һ�����ӵ�17%ʱ�����ƽ�һ����չ��������������֮���ƽ�С����ͬʱ����������չ�ķ����Ϻ����Ƹ��������˸������䣬ͬʱ������FCC(ͼ����ɫԭ��)��HCP(ͼ�к�ɫԭ��)������١���Ӧ��ﵽ17.05%ʱ�����ƽ�һ������ʹ�����ٳ���ʧЧ����ʱ�н϶��BCC�ṹ����������FCC�ṹ�������١����Կ��������ٵı������ɾ���ȱ�������������������غɵ������½�һ����չ�������������Ƽ�����ķ��������մﵽʧЧ��

ͼ3 �����ٺͲ�ͬTBS�Ͼ����������ʧЧ����ͼ
Fig. 3 Tensile deformation failure processes of single crystal tungsten and twin tungsten with different TBS
ͼ3(b)��ʾΪTBSΪ1.09 nm���Ͼ����������ʧЧ���̡���Ӧ��ﵽ7.85%ʱ���Ͼ��紦��ʼ�������ƣ���Ӧ��ﵽ7.9%ʱ�����ƿ�ʼ���֮࣬ǰ���ֵ����ƿ�ʼ����ƽ�����Ͼ����(100)������չ��ͬʱ��������չ�ķ������м���������ԭ����BCC�����Ų���ʽת������FCC����Ӧ��ﵽ8%ʱ��֮ǰ��չ��һ����ģ�����Ʊ�����������һ����չ����һЩ��С�����Ƴ����ˡ����ϡ�������ԭ���γ�С���Ƶ�λ�ô�������ȥ�Ͼ���(Detwinning)����Ӧ���һ�����ӵ�8.05%ʱ�����������Ĵ����ƽ�һ����չʹ�Ͼ��ٳ���ʧЧ����ʧЧ��λҲ�������������HCP����������ǰ���ֵ�ȥ�Ͼ�������Ҳ�õ���һ����չ��
ͼ3(c)��ʾΪTBSΪ2.73 nm���Ͼ����������ʧЧ���̡���Ӧ��ﵽ6.8%ʱ���ڲ�ͬ���Ͼ����ϳ�����4�����ƣ������ֲ���ͬһ���Ͼ����ϣ����Ͼ�����ϳ��ֵ����Ƴ��ȴ����Ͼ��������ơ���Ӧ���ʴﵽ6.85%ʱ���������ƵĽ�һ����չ������δʧЧ������Ȼ�ܵ������غ����ã����ڵ��Ͼ�������ܵ��ֲ��������غ����ã��Ӷ������������ļ����������Ρ���Ӧ��ﵽ6.9%ʱ���Ͼ�����ϵ�������չ���˸�������ƣ����Ͼ�����ϵ�����Ҳ������ͼ3(b)�еġ����ϡ�������Ȼ�Ͼ���û�з����˻�����Ҳû�лָ����������̬������������չ�ķ���������������ԭ����BCC����ת������FCC����Ӧ���ʴﵽ7%ʱ�����Ƶ���չʹ�Ͼ��ٳ���ʧЧ���Ͽڷdz���ƽ�����������ѵIJ�λ�в����Ĵ���λ�������������������Ͼ������������Ҳ�����ˣ��Ҷ������ڿ�ҪʧЧ���ѵ�ʱ���ֵ�λ��Ҳʮ�����ƣ�������������ʧЧλ�õĸ������������������ᵽ�ļ����������ε��µļ���Ӧ����Ϊλ���κˡ����Ƶ���������
ͼ3(d)��ʾΪTBSΪ5.48 nm���Ͼ����������ʧЧ���̡������ʧЧ���̷dz�����Ӧ��ﵽ6.05%ʱ�������ƣ���������ƽ���ھ���ķ���һֱ��չ��ʧЧ����ʧЧӦ��ֻ��6.2%��ʧЧ������û�г�����䣬����û�г��ֶ�������ơ�
������ķ������Կ����������ٵ�������չ����Ϊ(210)�����Ͼ��������Ƶ���չ����ƽ�����Ͼ��磻�ҵ����������Ƴ��ֵ�λ��û�й��ɣ����Ͼ��������е����ƶ��������Ͼ����С������ٱ���ʧЧ�����г����˽϶����䣬��Ҫ��BCCת��ΪFCCΪ�������Ͼ����У��������TBS�����Ӷ����٣�TBS��С���Ͼ����ڱ���ʧЧ�Ĺ����л��������Ͼ���ı��κ�ȥ�Ͼ��������Ͼ���ԽС�Ͼ���ı��κ�ȥ�Ͼ���Խ�࣬�Ͼ��ٵĶ���Ӧ��Ҳ��֮���ӡ����Կ�����������ʧЧ�����У��������Ͼ��ٻ����е���䡢�Ͼ���ı����Լ�ȥ�Ͼ�����������Ч�������Ͼ��ٵ�ʧЧӦ�䡣�������������������չ�ļ�ˣ��������Ƶ���ò��������չ�ķ�����һ����Ӱ�졣�Ͼ���ı��κ�ȥ�Ͼ������»���������������ܵ���һ���ļ���Ӧ����ʹ����ʧЧ�����г��ִ���λ�����ı������Ƶ���״��������������չ���ٶȣ��Ӷ�������Ͼ��ٵ�ʧЧӦ�䡣
ͼ4��ʾΪ�����ٺͲ�ͬ�Ͼ���������ι����в�ͬӦ���µ�ԭ�������ֲ�ͼ�����ܷ�ӳ�����ι����е���ԭ���ܵ�������ʩ�Ӹ�����������ͼ4(a)��ʾ��������������ǰ�����������ԭ�������ķֲ��dz����ȣ�������Ӧ������Ӷ����ӣ�ֱ����������ʱ�����������ߵ�ԭ�����ܵ�������ʼ���Ͳ���С��0 GPa������
��ͼ4(b)~(d)���Կ������Ͼ�����Ӧ��Ϊ0%ʱ��λ���Ͼ��紦��ԭ�Ӿ��ܵ��˽ϴ����������ͨ���Ŵ��ԭ��ϸ��ͼ���Կ������Ͼ������Ĺ���{010}����ԳƵ�3��ԭ���У�һ��ԭ���ܵ��ϴ����Ӧ��(ͼ�г�ɫԭ��)����������ԭ�����ܵ��ϴ��ѹӦ��(ͼ����ɫԭ��)����3��ԭ���ڲ���Ȼ����һ�����������������������ԭ�ӽṹ�����������Ͼ����У���˿�����Ϊ�Ͼ����д��ڽϴ����Ӧ�������Ͼ������нϸߵľ��索���ܡ�������������ʩ�������غ�ʱ���Ͼ��紦���������ϸߣ������ԭ���������Ѿ�����������γɿ�λ�ͼ�϶ԭ�ӵȵ�ȱ�ݣ������ȱ��ͬʱ�������Ͼ����ϣ�������չ�ɶ������ơ���ܺõؽ������Ͼ��ٵĴ����Լ���ʧЧӦ��ԶԶС�ڵ����ٵ�����ͬʱ����������ԭ�ӽṹ�ظ��������Ͼ����У�ʹ�Ͼ�����кܸߵľ����ܣ���Ϊ���κ���λ�����κ��뻬���ṩ��������ʹλ�������ھ����κˡ�
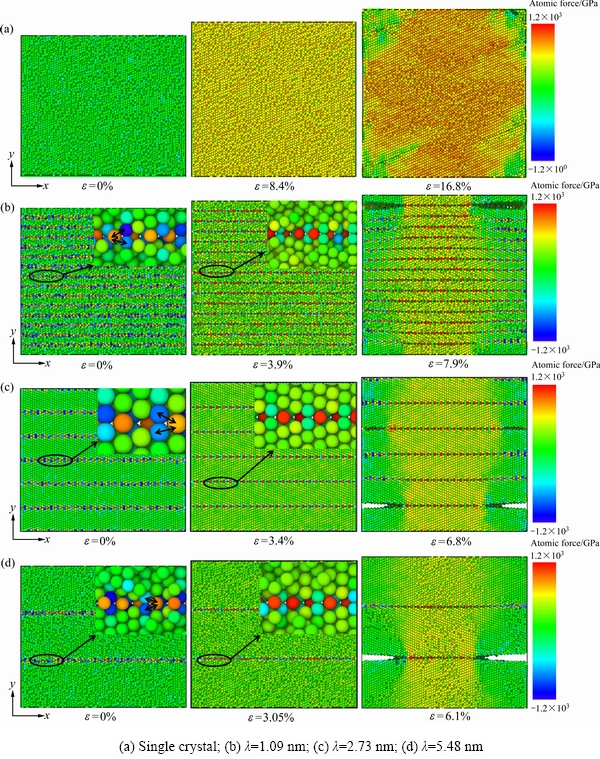
ͼ4 �����ٺͲ�ͬTBS�Ͼ���������ι����в�ͬӦ���µ�ԭ�����ֲ�ͼ
Fig. 4 Atomic force distribution of single crystal tungsten and twin tungsten with different TBS under different strain during tensile deformation
2.3 λ���ܶȶ����Ͼ�����ѧ���ܵ�Ӱ��
λ�����ھ����������غ�ʱ����ѧ�����зdz����Ӱ��[35]����ˣ�Ϊ���о�λ���Ե����ٺͲ�ͬTBS���Ͼ������ܵ��������غ�ʱ��Ӱ�죬����DXA�����������ʧЧ�����е�λ���߳��Ƚ�����ͳ�ơ�λ����ͳ���Ǵ�λ���ĵ�һ�γ��ֵ�������ȫʧЧ�Ĺ��̡�ͼ5��ʾΪ�����ٺ��Ͼ�����������ι�����λ���߳��ȵı仯��ͼ5(a)��ʾΪ��������������ι�����λ���ߵı仯�����Կ������α�����У�����������1/2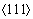 ȫλ������������Ӧ���16.7%���ӵ�16.8%�������ڲ�λ���κ˳�����ٶ�ԶԶ����λ��������ٶȣ�1/2ȫλ���ں̵ܶ�ʱ���ڿ�����������ֵ��ͬʱ
ȫλ������������Ӧ���16.7%���ӵ�16.8%�������ڲ�λ���κ˳�����ٶ�ԶԶ����λ��������ٶȣ�1/2ȫλ���ں̵ܶ�ʱ���ڿ�����������ֵ��ͬʱ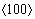 λ��Ҳ�ڿ������ӣ���ʱ�����ڲ���λ�������ڲ���Ͷѻ�����ʱ��Ӧ��Ӧ������Ҳ����ƽ������1/2λ����λ�����ﵽ��ֵ��λ���IJ���Ͷѻ�Ҳ�ﵽ�����Ӧ��Ӳ��[36-37]�����յ��²��ϵĶ���ʧЧ��
λ��Ҳ�ڿ������ӣ���ʱ�����ڲ���λ�������ڲ���Ͷѻ�����ʱ��Ӧ��Ӧ������Ҳ����ƽ������1/2λ����λ�����ﵽ��ֵ��λ���IJ���Ͷѻ�Ҳ�ﵽ�����Ӧ��Ӳ��[36-37]�����յ��²��ϵĶ���ʧЧ��
��ͼ5(b)~(d)���Կ��������Ͼ����У�1/2λ�����α�����г��ֵ���Ҫλ����TBSΪ2.73 nm���Ͼ����������α�����г��ֵ�1/2λ���߳�����С��Ϊ�����������ι�����1/2λ���߳��ȵļ�Сֵ���������Ͼ�����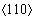 λ����λ�����ֳ�����˷����������ֵĹ��ɡ�������Ͼ��٣���������1/2λ���ߺ�λ���ߵij��ȶ�Ҫ���Ͼ��ٴ�һ����������������Ϊ�Ͼ����ڵ�λ�����Ͼ��紦�κˣ�Ȼ�����ڻ��������������Ƶ���һ���Ͼ��紦ʱλ�����˶����裻��������λ���Ļ����˶��������������⣬ֱ��λ�����Ƶ���������õ�ʱ��ſ�ʼֹͣ������
λ����λ�����ֳ�����˷����������ֵĹ��ɡ�������Ͼ��٣���������1/2λ���ߺ�λ���ߵij��ȶ�Ҫ���Ͼ��ٴ�һ����������������Ϊ�Ͼ����ڵ�λ�����Ͼ��紦�κˣ�Ȼ�����ڻ��������������Ƶ���һ���Ͼ��紦ʱλ�����˶����裻��������λ���Ļ����˶��������������⣬ֱ��λ�����Ƶ���������õ�ʱ��ſ�ʼֹͣ������
3 ����
1) �Ͼ��ٵĵ���ģ�������Ͼ�������Ӷ��������������ڵ����٣������ӵ��ٶ���TBSС��2.73 nmʱ�Ͽ죬��TBS����3.28 nm�����������Ҫԭ���������Ͼ���֮�������õ����Ͼ�������������
2) �Ͼ��ٵ�����������Ϊ��Ҫ���ɼ������Ͼ��紦�ı��Ρ����ѡ�����ȥ�Ͼ���������ɣ���������Ͼ��ٵ�����ǿ����Ҫȡ�����Ͼ���Ŀ������������Ͼ���ı��κ�ȥ�Ͼ������»����г��ִ���λ�����ı������Ƶ���չ��ʽ�Ӷ��������Ͼ���ı����������������Ͼ����ܶȵ����ӿɱ��ε��Ͼ������ӣ��Ͼ��ٵ�ʧЧӦ�����ӡ�
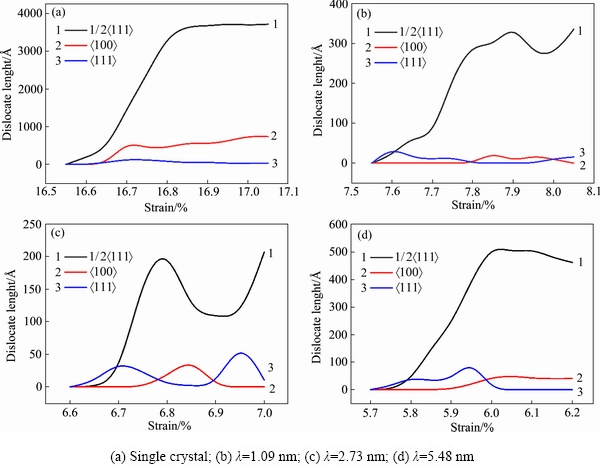
ͼ5 �����ٺͲ�ͬ�Ͼ���������ι�����λ���ߵij��ȱ仯����
Fig. 5 Variation curves of dislocation line length of single crystal tungsten and different twin tungsten during tensile deformation
3) �����Ͼ����д��������ϸߵ�����õ�������ԭ�ӽṹ��������ԭ�Ӵ��ڽϴ������ö��ﵽһ�������ϸߵ�ƽ��״̬��ʹ���������غ��º����ױ��ƻ����γɵ�ȱ�ݣ���һ���Ͼ����д��ڽ϶��ȱ��ʱ�������γ����ƣ����������غɵ���������չ���յ��¾�����ֶ���ʧЧ��
4) �����Ͼ���Ĵ��ڣ�λ���ھ����ڲ�����ʱ���ܵ��Ͼ�����谭��ֹͣ������������ֵ�������λ��������γ�Ӧ��Ӳ�����������Ͼ��ٵ�����ǿ��ԶС�ڵ����١�
REFERENCES
[1] MA Bin, RAO Qiu-hua, HE Yue-hui. Effect of crystal orientation on tensile mechanical properties of single-crystal tungsten nanowire[J]. Transactions of Nonferrous Metals Society of China, 2014, 24(9):2904-2910.
[2] �� Ƽ, ������, �� Ȫ, ��. ���ٸ�ѹŤת��֯�����ܵ��۳߶ȷ���[J]. ϡ�н��������빤��, 2019, 48(7): 2220-2224.
LI Ping, HUA Ya-ling, LIN Quan, et al. Microstructure evolution and micro-mechanical properties of pure tungsten during high pressure torsion[J]. Rare Metal Materials and Engineering, 2019, 48(7): 2220-2224.
[3] ������, ��һƽ, ������, ��. ���������ҹ��ٲ�ҵ��չ����̽��[J]. �й���ҵ, 2015, 24(1): 15-19, 51.
TAO Ying-long, FANG Yi-ping, WANG Zhen-zhen, et al. Discussion on the development of tungsten industry under the new situation in China[J]. China Mining Magazine, 2015, 24(1): 15-19, 51.
[4] CHU Ke, JIA Cheng-chang, LIANG Xue-bing, et al. Effect of powder mixing process on the microstructure and thermal conductivity of Al/diamond composites fabricated by spark plasma sintering[J]. Rare Metals, 2010, 29(1): 86-91.
[5] CHOI J, SUNG H M, ROH K B, et al. Fabrication of sintered tungsten by spark plasma sintering and investigation of thermal stability[J]. International Journal of Refractory Metals and Hard Materials, 2017, 69: 164-169.
[6] LI Xiao-qiang, HU Ke, QU Sheng-guan, et al. 93W-5.6Ni-1.4Fe heavy alloys with enhanced performance prepared by cyclic spark plasma sintering[J]. Materials Science and Engineering A, 2014, 599: 233-241.
[7] WEI Q, JIAO T, RAMESH K T, et al. Mechanical behavior and dynamic failure of high-strength ultrafine grained tungsten under uniaxial compression[J]. Acta Materialia, 2005, 54(1): 77-87.
[8] REISER J, RIETH M, DAFFEMER B, et al. Tungsten foil laminate for structural divertor applications��Basics and outlook[J]. Journal of Nuclear Materials, 2012, 423(1/3): 1-8.
[9] MATHAUDHU S N, DEROSSET A J, HARTWIG K T, et al. Microstructures and recrystallization behavior of severely hot-deformed tungsten[J]. Materials Science and Engineering A, 2008, 503(1): 28-31.
[10] BEAVEN P A, SMITH D A, MILLER M K, SMITH G D W. Combined FIM/TEM determination of the structure of an incoherent twin boundary in tungsten[J]. Philosophical Magazine A, 1981, 43(5): 1063-1070.
[11] MIKHAILOVSKIJ I M,WANDERKA N, KSENOFONTOV V A, et al. The �� structure of the lateral twin boundary in tungsten[J]. Philosophical Magazine Letters, 2007, 87(10): 743-750.
[12] WENSKY D A, WENSKY A K. Electronic structure of the tungsten pentacarbonylamine molecules[J]. Spectrochimica Acta Part A Molecular Spectroscopy, 1975, 31(1): 23-28.
[13] KRASKO G L. Effect of impurities on the electronic structure of grain boundaries and intergranular cohesion in tungsten[J]. International Journal of Refractory Metals and Hard Materials, 1993, 12(5): 251-260.
[14] �����, �ι���, �� ��, ��. �Ͻ�Ԫ�ؼ��ڶ�����ٵ�Ӱ��[J]. ����Ϲ���, 1998(6): 13-15.
WANG Yu-jin, SONG Gui-min, ZHOU Yu, et al. Effect of alloying elements and second phase on tungsten[J]. Aerospace Materials and Technology, 1998(6): 13-15.
[15] ZHANG Zhao-hui, WANG Fu-chi. Research on the deformation strengthening mechanism of a tungsten heavy alloy by hydrostatic extrusion[J]. International Journal of Refractory Metals and Hard Materials, 2001, 19(3): 177-182.
[16] LI J, ZHANGJ Y, LIU G, et al. Using the room temperature creep to strengthen nanotwinned Ni: The scaling behavior between the twin thickness and the grain size[J]. Materials Today Nano, 2020, 11: 100086.
[17] XU S, CHAVOSHI S Z, SU Y. Deformation mechanisms in nanotwinned tungsten nanopillars: Effects of coherent twin boundary spacing[J]. Physica Status Solidi (RRL)�CRapid Research Letters, 2018, 12: 1700399.
[18] WANG Yong, YU Wei-dong, WANG Fu-mei. Strand-spacing dependency on the tensile response of tri-component elastic-conductive composite yarns[J]. Textile Research Journal, 2018: 004051751876715.
[19] ��껷�, ������. ���÷��Ӷ���ѧ�о��ݶ������Ͼ�Cu���۱��λ���[J]. ����ѧ��, 2014, 50(2): 226-230.
ZHOU Hao-fei, QU Shao-xing. Investgation of atomistic deformation mechanism of gradient nanotwinned copper using molecular dynamics simulation method[J]. Acta Metallurgica Sinica, 2014, 50(2): 226-230.
[20] ���캵, �� ��. �Ͼ���Ԧ�����ѧ����Ӱ���ģ���о�[J]. ���ղ���ѧ��, 2017, 37(1): 73-79.
XU Tian-han, HE Song. Simulation of effect of twin boundary on mechanical property of ��-Fe[J]. Journal of Aeronautical Materials, 2017, 37(1): 73-79.
[21] �� ��, �� ��, �� ��, ��. þ�Ͻ���{1012}��{1011}�����Ͼ�����Ŀɶ��ԱȽ�[J]. �й���ɫ����ѧ��, 2019, 29(3): 508-516.
LI Heng, LIU Zhao, ZHANG Zen, et al. Mobility comparison of twin interfaces between {1012} and {1011} for magnesium alloy[J]. The Chinese Journal of Nonferrous Metals, 2019, 29(3): 508-516.
[22] �ι�ʤ, ţ��ά, ��ʿ��, ��. þ�Ͻ����Ťת���ε��Ͼ�����[J]. �й���ɫ����ѧ��, 2020, 30(7): 1574-1583.
SONG Guang-sheng, NIU Jia-wei, ZHANG Shi-hong, et al. Twinning mechanism of magnesium alloy rod torsion[J]. The Chinese Journal of Nonferrous Metals, 2020, 30(7): 1574-1583.
[23] �� ��, ��С��, ����ɽ, ��. һ�ּ���LAMMPS���Ӷ���ѧ�����IJ������������淶[J]. ������Ϣ��������Ӧ��, 2018, 9(4): 3-14.
YU Chao, YANG Xiao-yu, ZHAO Xu-shan, et al. A parameter description method and specification for integrated LAMMPS molecular dynamics software[J]. E-science Technology and Application, 2018, 9(4):3-14.
[24] SAINATH G, CHOUDHARY B K, JAYAKUMAR T. Molecular dynamics simulation studies on the size dependent tensile deformation and fracture behaviour of body centred cubic iron nanowires[J]. Computational Materials Science, 2015, 104: 76-83.
[25] ���, �� ��, ������, ��. ���Ӷ���ѧ�о�He���٦�3{112}�Գƾ����������ܵ�Ӱ��[J]. �����ȴ���ѧ��, 2019, 40(8): 96-104.
LI Fang-biao, GAO Ning, LI Yi-peng, et al. Effect of helium on tensile properties of tungsten ��3{112} symmetrical grain boundary studied by molecular dynamics[J]. Transactions of Materials and Heat Treatment, 2019, 40(8): 96-104.
[26] ��Դ��, �����, �� ��. ����/�ྦྷ����������й��̷��Ӷ���ѧģ��[J]. �й���ɫ����ѧ��, 2020, 30(8): 1837-1845.
LI Yuan-cai, JIANG Wu-gui, ZHOU Yu. Molecular dynamics simulation on shear mechanical properties of single crystal/polycrystalline Ni composites[J]. The Chinese Journal of Nonferrous Metals, 2020, 30(8): 1837-1845.
[27] ������, �� ��, �� ��, ��. Al2Cu������εķ��Ӷ���ѧģ��[J]. �й���ɫ����ѧ��, 2018, 28(9): 1746-1754.
LIU Xiao-bo, XIONG Zhen, FANG Zhou, et al. Molecular dynamics simulation of tensile deformation of Al2Cu[J]. The Chinese Journal of Nonferrous Metals, 2018, 28(9): 1746-1754.
[28] �� ��, ʱ����, �� ��. ��-Fe�к����谭λ���ƶ�Ӳ���ķ��Ӷ���ѧ�о�[J]. ԭ�����������ѧ��, 2018, 35(6): 1029-1036.
LIU Xing, SHI Jing-yi, PENG Lei. Molecular dynamics study of hardening induced by helium bubbles impeding edge dislocation motion in ��-Fe[J]. Journal of Atomic and Molecular Physics, 2018, 35(6): 1029-1036.
[29] ZHOU X W, WADLEY H N G, JOHNSON R A, et al. Atomic scale structure of sputtered metal multilayers[J]. Acta Materialia, 2001, 49(19): 4005-4015.
[30] SHU X T, XIAO S F, DENG H Q, et al. Atomistic simulation of crack propagation in single crystal tungsten under cyclic loading[J]. Journal of Materials Research, 2017, 32(8): 1474-1483.
[31] SWOPE W C, ANDERSEN H C, BERENS P H, et al. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters[J]. Journal of Chemical Physics, 1982, 76(1): 637-649.
[32] DING J, WANG Lu-sheng, SONG Kun, et al. Molecular dynamics simulation of crack propagation in single-crystal aluminum plate with central cracks[J]. Journal of Nanomaterials, 2017, 2017: 1-12.
[33] STUKOWSHI A. Visualization and analysis of atomistic simulation data with OVITO��The Open Visualization Tool[J]. Modelling and Simulation in Materials Science and Engineering, 2010, 18(1): 2154-2162.
[34] CHEN D, KULKARNI Y. Entropic interaction between fluctuating twin boundaries[J]. Journal of the Mechanics and Physics of Solids, 2015, 84: 59-71.
[35] CAO Yang, NI Song, LIAO Xiao-zhou, et al. Structural evolutions of metallic materials processed by severe plastic deformation[J]. Materials Science and Engineering R, 2018, 133: 1-59.
[36] HUANG Ling, LI Qing-jie, SHAN Zhi-wei, et al. A new regime for mechanical annealing and strong sample-size strengthening in body centred cubic molybdenum[J]. Nature Communications, 2011, 2: 547.
[37] GREER J R, WEINBERGER C R, CAI W. Comparing the strength of f.c.c. and b.c.c. sub-micrometer pillars: Compression experiments and dislocation dynamics simulations[J]. Materials Science and Engineering A, 2007, 493(1/2): 21-25.
Molecular dynamics simulation of effect of twin boundaries spacing on mechanical properties of nano-tungsten
XUE Ke-min, ZHANG Yong-qiang, WANG Lu-sheng, YAN Si-liang, LI Ping
(School of Materials Science and Engineering, Hefei University of Technology, Hefei 230009, China)
Abstract: In order to study the effect of twin boundaries spacing on the mechanical properties and deformation mechanism of nano-tungsten, uniaxial tensile simulation of twin tungsten with different twin boundaries spacing was carried out by molecular dynamics. The deformation failure process and microstructure evolution of nano-tungsten during tension were characterized and analyzed by using common neighbor analysis(CNA) and dislocation analysis method (DXA).The micro mechanism of the influence of twin boundaries spacing on the mechanical properties of nano tungsten was revealed. The results show that the phase transformation, the deformation of twin boundary and the phenomenon of de-twinning in the process of twin tungsten deformation will change the mode of crack propagation in twin tungsten and improve the deformation ability of twin boundary. With the decrease of twin boundaries spacing, that is, the increase of twin density, the number of deformable twin boundaries increases, resulting in the increase of fracture strain of nano-twin tungsten. Due to the existence of a special triatomic structure with high energy interaction in the twin boundaries, crystal defects are more likely to appear in the nano tungsten. The defects will form cracks quickly under the tensile load, resulting in the crystal fracture failure and seriously reducing the yield strength of the nano tungsten. Additionly, the existence of twin boundaries significantly reduces the number of geometric necessary dislocations and hinders the slip movement of dislocations, which makes it difficult for dislocations to emit and move, resulting in poor plasticity.
Key words: tungsten; nano twin boundary; twin distance; dislocation; molecular dynamics
Foundation item: Projects(51975175, 51675154) supported by the National Natural Science Foundation of China
Received date: 2020-08-24; Accepted date: 2020-12-03
Corresponding author: LI Ping; Tel: +86-551-62901368; E-mail: li_ping@hfut.edu.cn
(�༭ ��ѧ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(51975175��51675154)
�ո����ڣ�2020-08-24�������ڣ�2020-12-03
ͨ�����ߣ��� Ƽ�����ڣ���ʿ���绰��0551-62901368��E-mail��li_ping@hfut.edu.cn

