DOI:10.19476/j.ysxb.1004.0609.2018.07.22
硫化矿浮选捕收剂分子结构与性能的电子态密度研究
卢绿荣1,陈建华1, 2,李玉琼1
(1. 广西大学 资源环境与材料学院,南宁 530000;
2. 广西大学 金属资源高效利用及环境保护协同创新中心,南宁 530000)
摘 要:采用电子态密度研究了黄药、黑药和硫氨酯等硫化矿浮选捕收剂的电子结构与性能的关系。结果表明:电子态密度的离域性和轨道杂化作用较好地反映浮选药剂捕收性和选择性的强弱;黄药分子中双键硫原子的态密度最活跃,而黄药离子中单键硫原子的态密度最活跃;直链黄药(C2~C5)的态密度随烃基碳链的增长逐渐增强;丙基黄药和丁基黄药异构体的态密度强于相应的正构体的态密度,而正戊基黄药的态密度稍强于异戊基黄药的态密度;黑药中3418A的电子态密度相对最大,表现出更强的电子活性;不同结构的硫氨酯其氮原子的态密度变化比硫原子的大,这有利于改善硫氨酯捕收剂的螯合能力。
关键词: 浮选药剂;态密度;离域性;轨道杂化
文章编号:1004-0609(2018)-07-1482-09 中图分类号:TD923 文献标志码:A
随着矿产资源的日渐贫、细、复杂化,难选矿产资源增多,同时国家对环境保护日益重视,矿物浮选需要更加高效的药剂来实现清洁生产,从而提高资源综合利用率。浮选药剂的性能与其分子结构有关,研究浮选药剂结构与性能的关系一直受到国内外学者的高度重视, 王淀佐[1-5]采用等张比容法、CMC法、HLB法、基团电负性等方法对浮选药剂分子和结构进行参数化,提出了浮选药剂亲水疏水性指数,从理论上建立了浮选药剂分子结构模型。见百熙[6]采用蒋明谦《高等药物化学》中的6个原理分析了浮选药剂分子结构及其规律。王淀佐等[7]采用分子碎片法和分子连接性指数研究了浮选分子结构与性能的关系。印万忠等[8]采用酸碱软硬度的势标度来表示药剂的结构和性能。周国华等[9]提出用化学反应电子转移数来表示捕收剂的分子结构和性能。NATARAJAN等[10]研究了17种羟肟酸浮选某铜锌矿的浮选分离系数和土壤有机碳吸附常数的关系,发现二者具有正相关。冯其明等[11]采用油水分配系数研究了几种脂肪酸浮选钛铁矿的性能。在量化参数研究方面,王淀佐[5]采用分子轨道理论研究了捕收剂分子结构与浮选性能的关系,孙传尧等[12]采用前线轨道理论和软硬酸碱理论等研究了捕收剂非极性基结构与性能的关系。孙伟等[13]采用前线轨道理论对不同结构的黄铜矿捕收剂分子进行计算,建立了黄铜矿捕收剂分子结构与选择性的关系[13]。刘广义等[14]采用密度泛函理论研究了氮原子对硫氨酯捕收剂选择性的影响。SARVARAMINI等[15-16]采用密度泛函理论研究了硫代磷酸浮选捕收剂结构和性能的关系。这些研究试图将捕收剂分子结构参数化,建立捕收剂分子物性指数与结构性能的关系,为浮选药剂开发提供依据;然而这些研究忽略了电子的精细结构和轨道杂化作用的影响,无法深入细致的分析捕收剂分子空间结构对其电子性质和吸附活性的影响。
捕收剂分子与硫化矿物表面的作用以化学作用为主,具有电子作用特征。捕收剂分子中电子和轨道的精细结构决定了捕收剂分子与矿物表面的作用构型和作用强度,另外硫化矿物一般含有过渡金属或重金属原子,这些金属原子容易与捕收剂分子形成配位作用,而捕收剂的配位作用与其分子轨道杂化有关。态密度(Density of states,DOS)描述了能量为E附近每单位能量间隔内的量子态数称为状态密度,是描述固体中电子运动状态最重要的一个参数,在固体物理、表面科学和界面吸附中获得广泛的应用[17-18]。态密度能够获得详细的电子结构,如轨道组成、轨道杂化、电子的局域性和离域性、轨道成键和反键作用等[19],这些性质对于分析浮选药剂的化学性质和吸附活性非常重要。陈晔等[20]采用态密度研究了空间结构对硫化矿物表面性质的影响,LONG等[21]采用态密度研究了水分子对方铅矿、闪锌矿表面吸附硫醇捕收剂的影响,但采用态密度来研究浮选药剂分子性质与性能的关系还未见报道。本文作者采用态密度理论研究了常见硫化矿捕收剂分子的分态密度结构,解释了捕收剂分子结构对键合原子轨道杂化作用的影响,揭示了捕收剂分子结构与电子性质的内在关系,对于新型高效捕收剂的开发具有重要意义。
1 实验
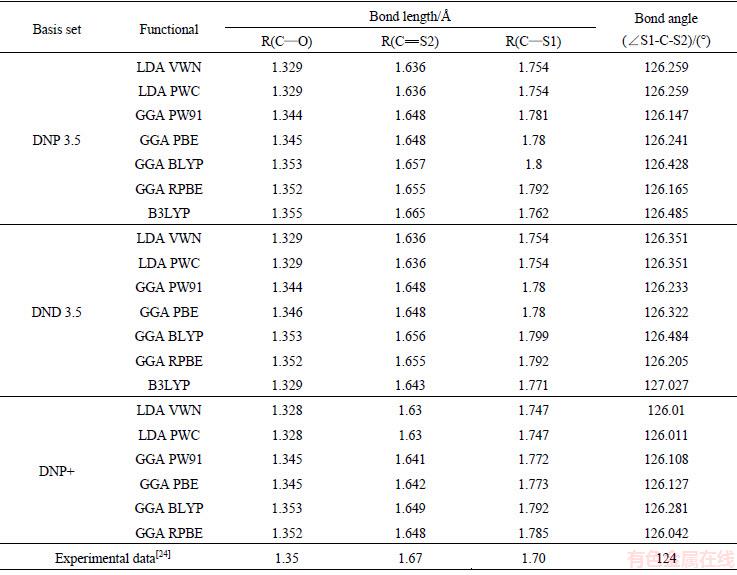
采用DMol3软件在密度泛函理论框架下[22-23],对硫化矿浮选捕收剂分子进行几何结构优化后计算电子态密度。本研究中对分子进行优化计算采用自旋极化,正式自旋为初始。在几何优化的收敛精度标准中,能量改变的收敛阈值为2.7×10-4 eV/atom,原子间作用力的收敛阈值为0.54 eV・nm,原子位移的收敛阈值为0.005 nm;自洽迭代收敛精度为1.0×10-6 eV/atom;原子自旋状态中多极展开为八极,密度混合电荷为0.2,密度混合自旋值为0.5,DIIS大小为6,拖尾效应值为0.1 eV。所有原子计算选取全电子。几何构型优化中泛函和基组的选择,通过采用不同的泛函和基组分别对乙基黄药分子构型进行优化,得到不同泛函和基组下乙基黄药分子(见图1)中碳硫双键C=S(S2)、碳硫单键C―S(S1)和碳氧键C―O的键长以及S=C―S键角,将表1中各个计算结果与文献值(1.70 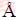 、1.67 和1.35 ,124°)进行[24]对比,得到GGA-RPBE泛函和DNP3.5基组下的值与文献值最为接近,从而确定几何构型优化中的泛函和基组为GGA-RPBE DNP3.5。对态密度的计算采用与几何优化相同的参数。
、1.67 和1.35 ,124°)进行[24]对比,得到GGA-RPBE泛函和DNP3.5基组下的值与文献值最为接近,从而确定几何构型优化中的泛函和基组为GGA-RPBE DNP3.5。对态密度的计算采用与几何优化相同的参数。
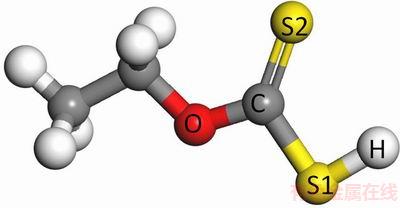
图1 乙黄药分子结构
Fig. 1 Molecular structure of ethyl xanthate
表1 乙黄药在不同泛函和基组下几何结构优化后的性质参数
Tabel 1 Properties of ethyl xanthate in different functionals and basis sets under the geometric optimization
2 结果与讨论
2.1 黄药类捕收剂
黄药是应用最广泛也是最重要的一类硫化矿捕收剂,其结构式为ROCSSM,在酸性溶液中极不稳定,在碱性条件下,黄药性质相对稳定。目前,我国选矿药剂厂生产的黄药产品以乙基黄药、丁基黄药和异丁基黄药为主,同时也生产(异)丙基黄药、(异)戊基黄药等。黄药几乎应用于金属硫化矿浮选的各个领域,是一种广谱性特征十分明显的硫化矿捕收剂[25]。
2.1.1 黄药类捕收剂中键合原子的确定
黄药分子的极性基团中含有氧、碳和硫原子,为了确定黄药分子中的键合原子,对乙黄药分子和离子分别进行电子态密度的计算,计算参数与几何结构优化参数一致。计算结果如图2所示。
态密度反映了电子在特定能级处的分布,一般而言,低能级的态密度表示电子相对稳定,高能级的态密度表示电子不稳定,活性较强。从图2可见,乙黄药分子和离子中的氧原子和碳原子的态密度都比硫原子更负,并且在费米能级(EF)附近,氧原子和碳原子的态密度都非常小,甚至接近于零,而硫原子在费米能级附近的态密度较大,费米能级附近的态密度代表了电子的活性大小,因此,黄药分子和离子中硫原子的反应活性最强,硫原子是黄药与硫化矿物表面作用的键合原子。这与前线轨道研究结果一致。
对于黄药分子而言,在费米能级附近的-2~0.5 eV,单键硫(S1)和双键硫(S2)的态密度不同(见图2(a)),这是由于单键硫原子与氢原子相连的缘故;双键硫在费米能级处的态密度大于单键硫,表明双键硫原子的电子活性比单键硫原子更强。在-8~-2 eV之间黄药分子的单键硫(S1)和双键硫(S2)的态密度也不相同,其中,双键硫原子在-6 eV附近发生了较强的s、p轨道杂化作用,而单键硫原子的轨道杂化作用较弱;根据研究结果[21],硫原子与过渡金属的成键作用主要发生在-8~-2 eV之间,因此双键硫原子的杂化作用能够促进黄药分子与硫化矿物表面作用。文献[11]结果表明,黄药分子的键合原子是双键硫原子,与态密度的理论分析结果一致。
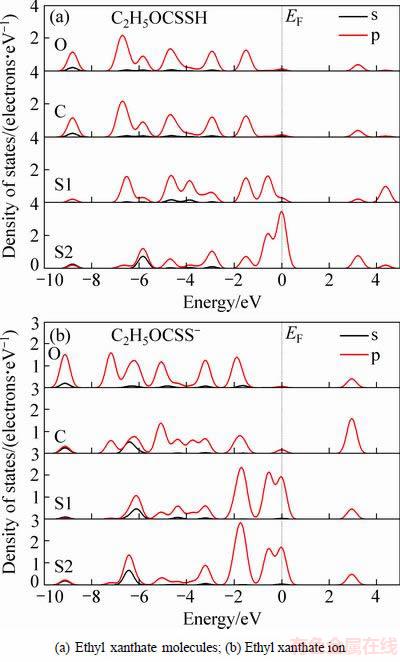
图2 乙黄药分子和乙黄药离子极性基团的电子态密度
Fig. 2 Electronic states density of polar groups of ethyl xanthate molecules and ethyl xanthate ion
对于黄药离子,其电子态密度与黄药分子有较大区别。首先,黄药离子在-2.5~0.5 eV之间的单键硫(S1)和双键硫(S2)的态密度几乎相同(见图2(b)),这是由于C―S单键和C=S双键产生了共振态,表明黄药离子的两个硫原子具有相近的电化学活性,研究结果表明黄药离子的两个硫原子都具有吸附活性[21]。另外,在-6~-3 eV之间单键硫原子的离域性比双键硫原子的强,有利于单键硫原子与硫化矿物表面的成键作用,模拟计算结果也表明单键硫原子与矿物表面成键的作用距离更小[21]。
比较图2(a)和(b)可知,从费米能级处硫原子的态密度来看,黄药离子的活性明显强于黄药分子的,黄药离子态密度离域性增强,因此黄药离子的反应活性大于相应的黄药分子的,这与文献[12]研究结果一致。
2.1.2 碳链长度对黄药捕收剂活性的影响
黄药的烃基碳链长度对黄药捕收性能具有显著影响,为了考察碳链长度与捕收性能之间的关系,分别计算了不同碳链长度的正构体黄药离子的态密度,结果如图3所示。
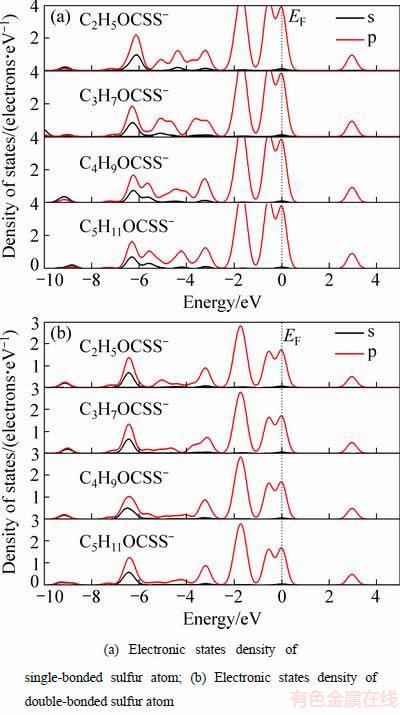
图3 不同碳链长度黄药中的硫原子态密度
Fig. 3 Electronic states densities of sulfur atoms in xanthates of different alkyl chain
从分态密度(见图3(b))可见,不同碳链长度下黄药中双键硫原子的态密度变化很小,说明碳链长度对双键硫原子的影响很小。从图3(a)可见,虽然在费米能级附近(-2.5~0.5 eV),碳链长度不影响黄药中单键硫原子的态密度,但在成键区-7.5~-2.5 eV之间,不同碳链长度黄药的单键硫原子态密度有明显区别,随烃基碳链的增长,黄药单键硫原子s、p轨道杂化作用逐渐增强,这也许就是长碳链黄药更容易与金属离子发生作用的原因。从以上分析可见,烃基长度对黄药捕收性能的影响主要是通过单键硫原子来实现的,碳链越长,单键硫原子的轨道杂化作用越强,越有利于黄药与金属离子的螯合作用。
2.1.3 同分异构体对黄药捕收剂活性的影响
丙基黄药、丁基黄药、戊基黄药的正异构体态密度计算结果表明,烃基的空间结构对黄药的影响类似于烃基长度的影响,即正异构体黄药的双键硫原子态密度相近,而单键硫原子的态密度不同。图4(a)~(c)显示了不同碳链长度的黄药正异构体中单键硫原子的态密度。
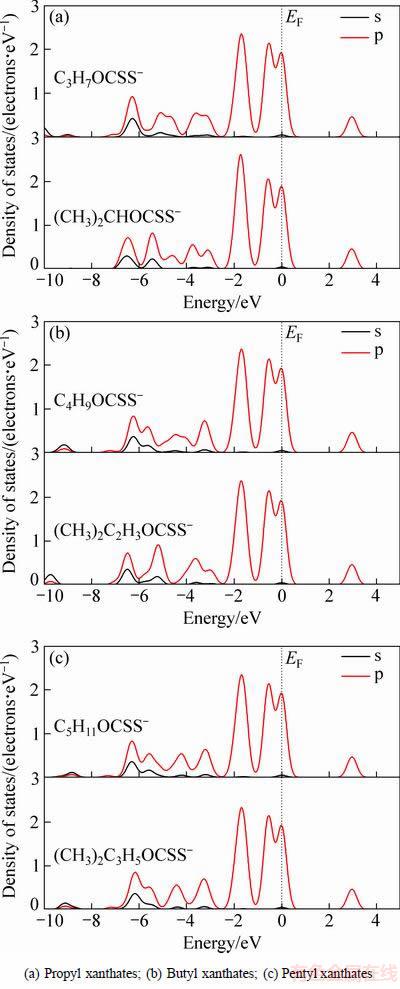
图4 黄药正异构体中单键硫原子态密度
Fig. 4 Electronic states densities of single-bonded sulfur atom in xanthate isomers
由图4可见,在费米能级附近(-2.5~0.5 eV),不同烃基结构的黄药中单键硫原子态密度是相同的,烃基结构对黄药单键硫原子的影响主要在-7~-3 eV能级区域。由图4(a)可见,异丙基黄药中单键硫原子的s、p轨道重叠较正丙基黄药大,杂化作用更强,且异丙基黄药在-5.5 eV附近出现了很强的杂化峰,在-4 eV处,异丙基黄药的态密度明显大于正丙基黄药的,由此说明异丙基黄药的单键硫原子活性比正丙基黄药的强。由图4(b)可知,异丁基黄药的单键硫原子的s、p轨道出现了3个明显的杂化峰,尤其在-6~-5 eV之间杂化作用强烈,与正丁基黄药相比,在-8~-2 eV区间杂化作用更强,说明异丁基黄药的单键硫原子活性比正丁基黄药的强。因此,异构体的丙基黄药和丁基黄药都比相应的正构体黄药捕收性更强。
然而烃基结构对戊基黄药的影响比对丙基黄药和丁基黄药要弱,在图4(c)中,正戊基黄药和异戊基黄药中单键硫原子态密度差异很小,但仍然可以发现,在-5.5 eV处正构体单键硫原子的杂化比异构体的强,出现了明显的杂化峰,说明正戊基黄药中的单键硫原子活性比异戊基黄药的强。浮选试验结果[26]也表明正戊基黄药的捕收性比异戊基的强。
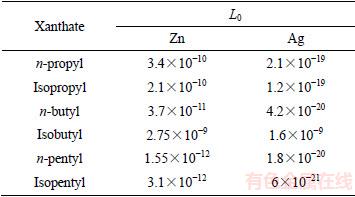
一般而言,键合原子的轨道杂化作用越强,就越容易与金属离子发生作用。从以上态密度讨论表明,异丙基和异丁基黄药的单键硫原子s、p轨道杂化作用强于相应正构体黄药,而异戊基黄药的单键硫原子s、p轨道杂化作用却弱于正戊基黄药。从表2不同黄药与锌和银的活度积数据可见,异丙基黄药和异丁基黄药与金属离子的活度积都比相应的正构体小,说明异丙基黄药和异丁基黄药与金属原子的作用强于相应正构体的;而异戊基黄药与金属离子的活度积却比正戊基黄药的大,说明异戊基黄药与金属离子的作用弱于正戊基黄药的。刘凤霞等[26]研究氧化铅的浮选结果表明异丙基黄药和异丁基黄药的捕收性能强于相应正构体,而异戊基黄药的捕收效果弱于正戊基黄药的。
表2 黄原酸锌和银的活度积L0[27]
Table 2 Relationship between activity product L0 of zinc and silver xanthate and alkyl structure and length[27]
2.1.4 3种烷基硫代碳酸浮选活性的比较
在乙黄药的基础上得到3种烷基硫代碳酸,即一硫代碳酸、二硫代碳酸和三硫代碳酸,分别计算单键硫原子的态密度,所得对比图如图5所示。
从图5可见,在成键区-8~-2 eV能级之间,一硫代碳酸、二硫代碳酸和三硫代碳酸中单键硫原子的s、p轨道的重叠逐渐增大,表明s、p轨道杂化作用逐渐增强,而且他们的态密度峰跨度也逐渐增大,表明离域性逐渐增强,即原子活性逐渐增强,同时,在此能级区间内,3种硫代碳酸单键硫原子的s、p轨道杂化峰逐渐增多,进一步说明原子活性逐渐增强,表现在浮选中对硫化矿的捕收能力逐渐增强,即三硫代碳酸对硫化矿的捕收性大于二硫代碳酸的,二硫代碳酸的捕收性大于一硫代碳酸的。
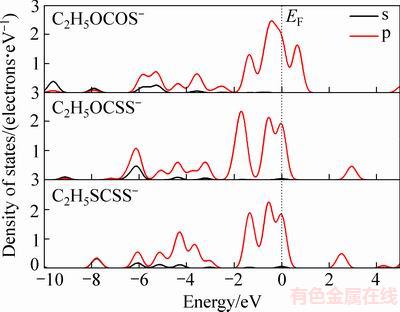
图5 不同硫代碳酸盐中键合硫原子的态密度
Fig. 5 Electronic states density of bonding sulfur atoms in different thiocarbonates
2.2 黑药类捕收剂
黑药类捕收剂的捕收能力比黄药稍弱,但选择性和稳定性强于黄药,具有较好的起泡性能[25]。甲酚黑药曾是各种黑药中应用最多的一种,对黄铜矿、方铅矿、闪锌矿具有较强的捕收能力,但甲酚黑药腐蚀性及毒性较大,有一定的污染,一般情况下应尽量少用。丁铵黑药是我国目前应用最为广泛的黑药类捕收剂,其选择性好,使用方便。苯胺黑药具有选择性好、捕收能力强等特点,对细粒方铅矿的捕收比甲酚黑药和乙基黄药更有效,可以在相对较低的pH值下实现铅锌硫和铜硫的分选。苯胺黑药和环己胺黑药对于氧化铅矿具有较好的捕收性能,用于混合铅锌矿和氧化铅锌矿的浮选时,其浮选指标优于用25号黑药和丁黄药作捕收剂的[25]。由美国氰特公司(Cytec)开发出来的二异丁基二硫代次磷酸钠(3418A)是一种新型的黑药类捕收剂,主要用于从高含量黄铁矿的矿石中浮选铅、铜和贵金属[28]。
对不同结构黑药中单键硫原子和双键硫原子的态密度研究结果表明,黑药类捕收剂的极性基团中具有较强的共轭效应,单键硫原子和双键硫原子的态密度几乎没有差异。图6所示为丁黑药((C4H9O)2PSS-)、甲酚黑药((CH3-C6H4-O)2PSS-)、环己胺黑药((C6H11-NH)2PSS-)、苯胺黑药((C6H5-NH)2PSS-)、3418A(((CH3)2C2H3)2PSS-) 5种黑药中单键硫原子的态密度。

图6 黑药类捕收剂分子中单键硫原子态密度
Fig. 6 Electronic states density of single-bonded sulfur atom in aerofloat collectors
由图6可见,在费米能级附近(-2~0.5 eV),甲酚黑药的态密度峰分裂程度不如丁黑药的,在-2 eV附近,甲酚黑药的态密度也比丁黑药的大,另外,在-7~-5 eV区间,苯胺黑药的s、p轨道的杂化作用也强于丁黑药的,说明甲酚黑药单键硫原子的活性比丁黑药的强,甲酚黑药表现出更好的捕收性和选择性。从环己胺黑药和苯胺黑药的单键硫原子态密度对比中也可以看出,苯胺黑药的离域性比环己胺黑药的强,尤其在费米能级附近(-2~0.5 eV)和-6~-4 eV区间,说明苯胺黑药的捕收性和选择性相对于环己胺黑药的更好。对于3418A而言,在费米能级附近,3418A的电子态密度和其他4种黑药的有明显不同,其态密度平缓,表现较强的离域性,且在-4~-2 eV区间,3418A的态密度相对最大,表现出更强的电子活性,说明3418A的捕收性和选择性比其他4种黑药的都强。
2.3 硫氨酯捕收剂
硫氨酯类捕收剂是硫化矿物浮选的优良捕收剂,属于非离子型极性捕收剂,其特点是选择性强、用量少,在酸性介质中比较稳定,有较强的起泡性,在弱碱性条件下表现出比黄药类捕收剂和黑药类捕收剂更好的选择性[25]。
乙硫氨酯(Z-200)是一种最有代表性的硫氨酯捕收剂,其学名为O-异丙基-N-乙基硫代氨基甲酸酯,鉴于氮原子上的取代烃基对硫氨酯捕收剂性能有显著影响,国内外研究开发出一些特殊结构的硫氨酯,即O-丁基-N-乙氧丙基硫代氨基甲酸酯(硫氨酯1)、O-烷基-N-烯丙基硫代氨基甲酸酯(硫氨酯2)和O-烷基-N-烷氧基羰基硫代氨基甲酸酯(硫氨酯3)[25]。
硫氨酯捕收剂分子中硫原子和氮原子都具有活性,分别计算乙硫氨酯及3种特殊结构的硫氨酯中硫原子和氮原子的态密度,如图7所示。
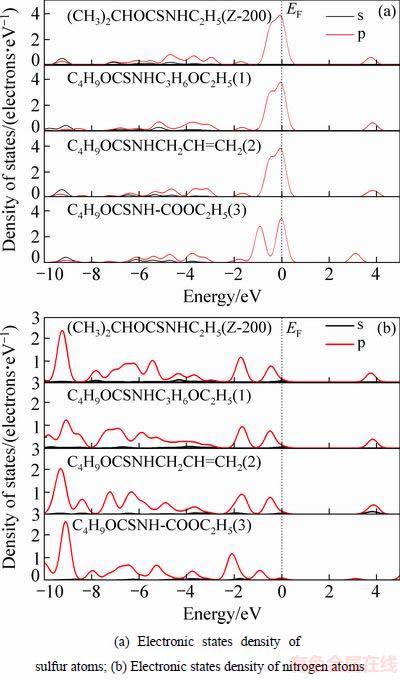
图7 硫氨酯捕收剂分子中硫原子和氮原子态密度
Fig. 7 Electronic states density of sulfur and nitrogen atoms in thocarbamate collectors
比较图7(a)和(b)可知,硫氨酯捕收剂中硫原子在费米能级处的态密度明显大于氮原子,同时在-7~-2 eV能级区间,硫原子的s、p轨道杂化作用也比氮原子的强,说明硫氨酯捕收剂中硫原子的活性比氮原子强。从图7(a)中可以看到,O-烷基-N-烷氧基羰基硫代氨基甲酸酯的硫原子态密度和其他3种硫氨酯有显著差异,其态密度离域性变弱,这是由于与氮原子相连接的烷基变成了羰基的缘故。同时从图7(b)中也能发现,O-烷基-N-烷氧基羰基硫代氨基甲酸酯的氮原子态密度也和其他三种硫氨酯不同,其态密度在费米能级附近变小,并发生负移,说明羰基的存在降低了硫氨酯捕收剂的活性。
对于乙硫氨酯和O-丁基-N-乙氧丙基硫代氨基甲酸酯(硫氨酯1)、O-烷基-N-烯丙基硫代氨基甲酸酯(硫氨酯2) 3种捕收剂而言,他们的氮原子和硫原子的态密度在费米能级附近几乎相同,而他们的差异主要表现在-8~-2 eV能级区间,这主要是由于硫氨酯结构中脂肪胺的烃基不同引起的,其中,氮原子的态密度变化比硫原子的大,这有利于改善硫氨酯捕收剂的螯合能力,因为氮原子比硫原子具有更强的配位能力。
3 结论
1) 利用电子态密度研究浮选药剂的结构与性能可以获得原子的电子分布和轨道杂化情况,有助于更好地判断浮选药剂的电子活性和键合能力,为研究浮选药剂的捕收性和选择性提供了有效可行的方法。
2) 黄药极性基团中硫原子的态密度最活跃,其中,黄药分子中双键硫原子的轨道杂化比单键硫原子的强,表现出更好的活性,而黄药离子中两个硫原子态密度相近,但单键硫原子的离域性更强;不同碳链长度下黄药双键硫原子的态密度变化很小,但单键硫原子态密度随烃基碳链的增长其硫原子s、p轨道杂化作用逐渐增强;在不同烃基结构的黄药中,异构体的丙基黄药和丁基黄药的态密度明显强于相应的正构体的,而正戊基黄药和异戊基黄药的态密度差异很小,且正戊基黄药的态密度强于异戊基黄药的;一硫代碳酸、二硫代碳酸和三硫代碳酸中键合硫原子的态密度逐渐增强。
3) 黑药中单键硫原子和双键硫原子的态密度几乎没有差异,甲酚黑药的态密度比丁黑药的大,苯胺黑药的态密度比环己胺黑药的大,而3418A的电子态密度和其他4种黑药的明显不同,表现出更强的离域性,说明3418A的态密度相对最大,表现出更强的电子活性。
4) 硫氨酯捕收剂中硫原子的态密度比氮原子的大,不同硫氨酯捕的态密度不同,这主要是由其结构中脂肪胺的烃基不同所引起的。
REFERENCES
[1] 王淀佐. 浮选药剂的结构与性能―浮选药剂的HLB计算法[J]. 有色金属(冶炼部分), 1977(7): 36-40.
WANG Dian-zuo. The structure and properties of flotation reagents―HLB calculation method of flotation reagents[J]. Nonferrous Metals (Smelting Part), 1977(7): 36-40.
[2] 王淀佐. 浮选药剂的结构与性能―浮选药剂性能的CMC计算法[J]. 有色金属(冶炼部分), 1977(6): 25-28.
WANG Dian-zuo. The structure and properties of flotation reagents―CMC calculation method of flotation reagents performance[J]. Nonferrous Metals (Smelting Part), 1977(6): 25-28.
[3] 王淀佐. 浮选药剂的结构与性能―浮选药剂性能的电负性计算法[J]. 有色金属(冶炼部分), 1975(4): 44-51.
WANG Dian-zuo. The structure and properties of flotation reagents―Electronegativity calculation method of flotation reagents performance[J]. Nonferrous Metals (Smelting Part), 1975(4): 44-51.
[4] 王淀佐. 浮选药剂的结构与性能―一百种含硫有机浮选剂的分子设计[J]. 有色金属(选矿部分), 1979(2): 14-28.
WANG Dian-zuo. The structure and properties of flotation reagents―Molecular design of one hundred kinds of sulfur-containing organic flotation reagents[J]. Nonferrous Metals (Concentrating Part), 1979(2): 14-28.
[5] 王淀佐. 矿物浮选和浮选剂[M]. 长沙: 中南大学出版社, 1986.
WANG Dian-zuo. Mineral flotation and flotation reagents[M]. Changsha: Central South University Press, 1986.
[6] 见百熙. 浮选药剂的分子结构及其规律性―用高等药物化学原理探讨浮选药剂分子的设计[J]. 有色金属工程, 1983(1): 10-18.
JIAN Bai-xi. Molecular structure and regularity of flotation reagents―A discussion on the design of flotation reagent molecules by the principle of higher medicinal chemistry[J]. Nonferrous Metals Engineering, 1983(1): 10-18.
[7] 王淀佐, 林 强, 蒋玉仁. 选矿与冶金药剂分子设计[M]. 长沙: 中南工业大学出版社, 1996.
WANG Dian-zuo, LIN Qiang, JIANG Yu-ren. Molecular design of mineral processing and metallurgy[M]. Changsha: Central South University of Technology Press, 1996.
[8] 王纪镇,印万忠. 酸碱软硬度的势标度在浮选剂结构性能研究中应用[J]. 东北大学学报(自然科学版), 2013, 34(7): 1035-1038.
WANG Ji-zhen, YIN Wan-zhong. Application of acid-base potential scale in structure performance study of flotation reagents[J]. Journal of Northeastern University(Natural Science), 2013, 34(7): 1035-1038.
[9] 周国华, 孙 伟, 薛玉兰, 蒋玉仁, 何伯泉. 化学反应电子转移数判据在浮选捕收剂结构与性能关系的应用研究[J]. 有色金属, 2001, 53(1): 19-21.
ZHOU Guo-hua, SUN Wei, XUE Yu-lan, JIANG Yu-ren, HE Bo-quan. Application of number of electronic transfer in chemical reaction to study of structure-reactivity relationship of flotation reagents[J]. Nonferrous Metals, 2001, 53(1): 19-21.
[10] NATARAJAN R,NIRDOSH I. Application of topochemical, topostructural, physicochemical and geometrical parameters to model the flotation efficiencies of N-arylhydroxamic acids[J]. International Journal of Mineral Processing, 2003, 71(1/4): 113-129.
[11] 冯其明, 席振伟, 张国范, 卢毅屏, 欧乐明. 脂肪酸捕收剂浮选钛铁矿性能研究[J]. 金属矿山, 2009(5): 46-49.
FENG Qi-ming, XI Zhen-wei, ZHANG Guo-fan, LU Yi-ping, OU Le-ming. Collecting performance of fatty acid as collector for ilmenite[J]. Metal Mine, 2009(5): 46-49.
[12] 曹 飞, 孙传尧, 王化军, 陈飞武. 烃基结构对黄药捕收剂浮选性能的影响[J]. 北京科技大学学报, 2014, 36(12): 1589-1594.
CAO Fei, SUN Chuan-yao, WANG Hua-jun, CHEN Fei-wu. Effect of alkyl structure on the flotation performance of xanthate collectors[J]. Journal of University of Science and Technology Beijing, 2014, 36(12): 1589-1594.
[13] 孙 伟, 杨 帆, 胡岳华, 何国勇, 刘文莉. 前线轨道在黄铜矿捕收剂开发中的应用[J]. 中国有色金属学报, 2009, 19(8): 1524-1530.
SUN Wei, YANG Fan, HU Yue-hua, HE Guo-yong, LIU Wen-li. Application of frontier orbital in developing new collectors of chalcopyrite[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(8): 1524-1530.
[14] LIU Guang-yi, ZHONG Hong, DAI Ta-gen, XIA Liu-yin. Investigation of the effect of N-substituents on performance of thionocarbamates as selective collectors for copper sulfides by ab initio calculations[J]. Minerals Engineering, 2008, 21(12/14): 1050-1054.
[15] SARVARAMINI A, LARACHI F. Understanding the interactions of thiophosphorus collectors with chalcopyrite through DFT simulation[J]. Computational Materials Science, 2017, 132: 137-145.
[16] LIU Guang-yi, XIAO Jing-jing, ZHOU Di-wen, ZHONG Hong, CHOI Phillip, XU Zheng-he. A DFT study on the structure-reactivity relationship of thiophosphorus acids as flotation collectors with sulfide minerals: Implication of surface adsorption[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2013, 434(19): 243-252.
[17] 张行荣, 刘崇峻, 朱阳戈, 郑桂兵, 尚衍波, 韩 龙, 艾 晶. 采用密度泛函方法研究铜铅分离的抑制机理[J]. 中国有色金属学报, 2017, 27(4): 843-849.
ZHANG Xing-rong, LIU Chong-jun, ZHU Yang-ge, ZHENG Gui-bing, SHANG Yan-bo, HAN Long, AI Jing. Depressing mechanism of copper-lead separation by density functional theory method[J]. The Chinese Journal of Nonferrous Metals, 2017, 27(4): 843-849.
[18] 张行荣, 郑桂兵, 艾 晶, 刘崇峻, 尚衍波. 赤铁矿反浮选淀粉抑制作用第一性原理[J]. 中国有色金属学报, 2016, 26(2): 465-470.
ZHANG Xing-rong, ZHENG Gui-bing, AI Jing, LIU Chong-jun, SHANG Yan-bo. First-principles of depressing mechanism of starch in reverse-flotation of hematite[J]. The Chinese Journal of Nonferrous Metals, 2016, 26(2): 465-470.
[19] 陈建华. 硫化矿物浮选固体物理研究[M]. 长沙: 中南大学出版社, 2015: 31-35.
CHEN Jian-hua. The solide physics of sulphide minerals flotation[M]. Changsha: Central South University Press, 2015: 31-35.
[20] 陈 晔, 陈建华, 李玉琼, 赵翠华. 空间结构对硫化矿物表面能带结构和电子性质的影响[J]. 中国有色金属学报, 2016, 26(11): 2403-2411.
CHEN Ye, CHEN Jian-hua, LI Yu-qiong, ZHAO Cui-hua. Effect of spatial structure on band structure and electronic properties of sulphide minerals[J]. The Chinese Journal of Nonferrous Metals, 2016, 26(11): 2403-2411.
[21] LONG Xian-hao, CHEN Ye, CHEN Jian-hua, XU Zheng-he, LIU Qing-xia, DU Zheng. The effect of water molecules on the thiol collector interaction on the galena (PbS) and sphalerite (ZnS) surfaces: A DFT study[J]. Applied Surface Science, 2016, 389: 103-111.
[22] JONES R O, GUNNARSSON O. The density functional formalism, its applications and prospects[J]. Reviews of Modern Physics, 1989, 61(3): 689-746.
[23] MARZARI N, VANDERBILT D, PAYNE M C. Ensemble density-functional theory for ab-initio molecular dynamics of metals and finite-temperature insulators[J]. Physical Review Letters, 1997, 79(7): 1337-1340.
[24] HUNG A, YAROVSKY I, RUSSO S P. Density-functional theory studies of xanthate adsorption on the pyrite FeS2(110) and (111) surfaces[J]. Journal of Chemical Physics, 2003, 118(13): 6022-6029.
[25] 孙传尧. 选矿工程师手册(第1册)[M]. 北京: 冶金工业出版社, 2015: 518-529.
SUN Chuan-yao. Handbook for mineral processing engineers[M]. Beijing: Metallurgical Industry Press, 2015: 518-529.
[26] 刘凤霞, 陈建华, 吴伯增, 朱文涛. 白铅矿、褐铁矿的硫化浮选试验研究[J]. 矿产保护与利用, 2007(1): 27-30.
LIU Feng-xia, CHEN Jian-hua, WU Bo-zeng, ZHU Wen-tao. Sulfidizing flotation tests of cerusite and limonite[J]. Conservation and Utilization of Mineral Resources, 2007(1): 27-30.
[27] B.A.格列姆博茨基. 浮选过程物理化学基础[M]. 郑飞, 译. 北京: 冶金工业出版社, 1985: 299-301.
GLEMBOTSKII В А. Physical chemistry basis of flotation process[M]. ZHENG Fei, transl. Beijing: Metallurgical Industry Press, 1985: 299-301.
[28] E.T.帕西纳-特雷维劳, 周廷熙, 李长根. 二异丁基二硫代次膦酸钠(Aerophine 3418A)与活化和未活化的方铅矿和黄铁矿的相互作用[J]. 国外金属矿选矿, 2004, 41(6): 20-26.
PASINA-TREVELLAO E T, ZHOU Ting-xi, LI Chang-gen. Interaction of activated and unactivated galena and pyrite with sodium diisobutyldithiophosphinate (Aerophine 3418A)[J]. Metallic Ore Dressing Abroad, 2004, 41(6): 20-26.
Electronic states density study of molecular structures and activity of sulfide floatation collectors
LU Lü-rong1, CHEN Jian-hua1, 2, LI Yu-jiong1
(1. School of Resources Environment and Materials, Guangxi University, Nanning 530000, China;
2. Innovation Center for Metal Resources Utilization and Environment Protection, Guangxi University, Nanning 530000, China)
Abstract: The density of electronic states method was used to study the relationship between structures and activity of sulfide floatation collectors such as xanthate, aerofloat and thocarbamate. The results show that delocalization and orbital hybridization could better reflect the collecting and selectivity of flotation reagents. The states density of double-bonded sulfur atom in xanthate is the most active, while that of single-bonded sulfur atom in xanthate ion. Moreover, the states density of straight chain xanthates (C2-C5) increases with the alkyl chain growth. The order of the states density of xanthate isomers is as follows: isopropyl xanthate>n-propyl xanthate, isobutyl xanthate>n-butyl xanthate and n-pentyl xanthate>isopentyl xanthate; The electronic states density of 3418A in aerofloat is relatively maximum, showing the stronger electron activity. Thocarbamate with different structures changes its states density of nitrogen atom more than that of sulfur atom, which is beneficial to improve the chelating ability of thocarbamate collectors.
Key words: flotation reagent; density of states; delocalization; orbital hybridization
Foundation item: Projects(51574092, 51364002, 51304054) supported by the National Natural Science Foundation of China; Project(2014GXNSFAA118316) supported by Guangxi Natural Science Foundation, China
Received date: 2017-01-20; Accepted date: 2017-07-20
Corresponding author: CHEN Jian-hua; Tel: +86-771-3232200; E-mail: jhchen@gxu.edu.cn
(编辑 龙怀中)
基金项目:国家自然科学基金资助项目(51574092,51364002,51304054);广西自然科学基金资助项目(2014GXNSFAA118316)
收稿日期:2017-01-20;修订日期:2017-07-20
通信作者:陈建华,教授,博士;电话:0771-3232200;E-mail:jhchen@gxu.edu.cn

