DOI: 10.11817/j.ysxb.1004.0609.2021-37885
��ϡ������ˮ����Re3+(H2O)n (n=1~12��Re=La��Ce��Pr��Nd)��DFT�о�
ŷ�Ҳ�1, 2��������1, 3��������4���Ⲯ��5������1
(1. ����������ѧ ��Դ�뻷������ѧԺ������ 341000��
2. ���ϡ�����Ź������ο�ҵͶ������˾������ 530022��
3. Ϋ������пҵ����˾��Ϋ�� 262100��
4. ����������ѧ ���������ѧԺ������ 341000��
5. �й���ҵ��ѧ(����) ��ѧ�뻷������ѧԺ������ 100083)
ժ Ҫ������Material studio��DMol3ģ���о���La3+��Ce3+��Pr3+��Nd3+�������ӵ�ˮ����Re3+(H2O)n (n=1~12)�������˼����Ż���Ĺ��͵ļ��νṹ������ܡ�ǰ�߹������ɡ�����������������La��Ce��Pr��Nd������ϡ������Re3+�ĵ�һˮ���㣬��Сˮ�����ֱ�Ϊ4��6��7��7����ͬˮ���������£�Re��Oƽ��������La��Ce��Pr��Nd��Re3+(H2O)10��Re��Oƽ������ԼΪ2.6 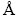 ����ϡ������ˮ�����ˮ����뾶ԼΪ3.22~3.59 ����ϡ�����ӵ�ˮ����ӦΪ���ȷ�Ӧ����ˮ������λ����ͬʱ������ܴ�С˳��Ϊ��La��Ce��Pr��Nd����ɷ��������ʾ��ˮ�Ϸ�Ӧ������ϡ�����ӵõ����ӣ�����һˮ����������Re3+(H2O)n�����������ȶ���Re���ӵĵ������2e���ҡ�ǰ�߹�����������ʾ����ͬ�����Ӱ뾶�ı仯������ϡ������������Ӳ��˳��Ϊ��La��Nd��P��Ce��ģ����La3+(H2O)n�ĺ�����ף�����ˮ�������������ӣ�Vsym��Vasym��������ƣ���n����10ʱ��Vsym��Vasym�岻�ٷ������ƣ�˵����һˮ�������ˮ����û����La���ӷ�����Ӧ��
����ϡ������ˮ�����ˮ����뾶ԼΪ3.22~3.59 ����ϡ�����ӵ�ˮ����ӦΪ���ȷ�Ӧ����ˮ������λ����ͬʱ������ܴ�С˳��Ϊ��La��Ce��Pr��Nd����ɷ��������ʾ��ˮ�Ϸ�Ӧ������ϡ�����ӵõ����ӣ�����һˮ����������Re3+(H2O)n�����������ȶ���Re���ӵĵ������2e���ҡ�ǰ�߹�����������ʾ����ͬ�����Ӱ뾶�ı仯������ϡ������������Ӳ��˳��Ϊ��La��Nd��P��Ce��ģ����La3+(H2O)n�ĺ�����ף�����ˮ�������������ӣ�Vsym��Vasym��������ƣ���n����10ʱ��Vsym��Vasym�岻�ٷ������ƣ�˵����һˮ�������ˮ����û����La���ӷ�����Ӧ��
�ؼ��ʣ�ϡ��������ˮ����ܶȷ�����Material studio
���±�ţ�1004-0609(2021)-xx- -���� ��ͼ����ţ�TD955���� ���ױ�־�룺A
���ĸ�ʽ��ŷ�Ҳ�, ������, ������, ��. ��ϡ������ˮ����Re3+(H2O)n(n=1~12��Re=La��Ce��Pr��Nd)��DFT�о�[J]. �й���ɫ����ѧ��, 2021, 31(x): xxxx-xxxx. DOI: 10.11817/j.ysxb.1004.0609.2021-37885
OU Jia-cai, ZHANG Tian-xi, HUANG Li-jin-hong, et al. DFT study on light rare earth ion hydrate Re3+(H2O)n (n=1~12, Re = La, Ce, Pr, Nd)[J]. The Chinese Journal of Nonferrous Metals, 2021, 31(x): xxxx-xxxx. DOI: 10.11817/j.ysxb.1004.0609.2021-37885
ϡ����������Ϊ��ҵά���أ��ڴ�����š���ѧ��ũҵ���Ƚ�������������Ҫ�㷺��Ӧ��[1]��������ȡ����ʱ�����IJ�ͬ�������������ϡ��Ԫ�ط�Ϊ��ϡ������ϡ������ϡ�����飺��ϡ��Ԫ��(LRe)������絽��(ԭ��������57��60)����ϡ��Ԫ����(MRe)�����̵���(ԭ��������62��64)����ϡ��Ԫ��(HRe)�������(ԭ������Ϊ65��71)������ɡ�
ϡ��������ˮ��Һ�лᷢ������ˮ����������ˮ����������������ѧ���������ѧ�ͻ�������������㷺��ע�����������ӽ���ˮ��Һ���γ��ȶ���ˮ�Ͻṹ�Ma+/b-(H2O)n[2-3]�����ȶ���ˮ�������о�ϡ�������ڸ���ʯ������������������������(����������ϡ�������)�������Լ�ϡ����ȡ�������Ҫ������������������ϡ��Ԫ�ص���Ҫ�Ļ�ѧ�����ԡ�����ĵ������ʺ�ѧ���ʣ�����ϡ�����ӵ�ˮ�Ͻṹ��Re3+(H2O)n�о�Խ��Խ�ܵ����ӣ��о����ֶ���Ҫ���������ģ�⣬����Ӷ���ѧģ��[4-7]�����ӻ�ѧģ��[6, 8-11]�ȣ������о�����XRD[12-16]��EXAFS[16-18]��XANES[19]��Raman[20-22]�ȣ��Լ�����ѧ�о�[23]�ȡ����еļ����ģ���о���Ҫ�о�1-2��ϡ�����ӵ�ˮ������û�н�ԭ�����������ϡ��������Ϊһ��������жԱ��о��������о�������ѧ�о��������ʾRe3+��H2O��ˮ���������ڽ���ܵļ��㲻��ȷ�������ܵ����������Ӱ�죬���¶ȡ�ˮ���������ӵȣ����⣬�����о���ˮ����������Ĺ��ױ仯û�н����۵Ľ��ͣ�����ѧ�о���ˮ����������Ľ���ܡ���ɡ����������Ϣ��ϵ������ͬ����ϡ��������ˮ��Һ�лᷢ������ˮ������Re3+(H2O)n���γɹ����ܵ�ˮ�ļ�������Ӱ�죬����ˮ������λ���������ӣ�ˮ����֮����ڿռ�λ��ЧӦ��ˮ������n����ʱ��(H2O)n��Re3+��ϵĸ����ܣ�ˮ��������n���ӣ�ˮ�����ܵ��˴˵��ų����ã�����λ���������ӣ����ջ��γ�һ�����ܵ�ˮ���Ӳ㣬����һˮ���㣻����ˮ���������ļ������ӣ�Reԭ����Χ��ʼ�γɵڶ����������ˮ���㡣�ӵ���ת�ƵĽǶȷ��������ڵ�һˮ����������ĵ�Reԭ�Ӹ�����������ԭ�ӵĵ���ת�Ƹ���ͻ������ˣ����Ľ��ص��о�Re3+(H2O)n�����һˮ��������ʣ��ڵ�һˮ������γɹ����У�����һ�������ٽ�ˮ������������λ��ˮ��������������ٽ�ֵʱ��Re3+(H2O)n��ʼ�γɵڶ�ˮ���㡣����ٽ�ֵ������һ�ɲ��䣬��ˣ�Re3+��H2Oˮ���γ�ˮ������һ����̬ƽ����̣�����ϡ���������ḻ�ĵ���������Ȼ�����£�����λ������9~12֮�䣬�����������ڹ�������ķ����г��֡�ϡ�����ӵ���ȡ������ѭ���ȷ����ٷ��롱�IJ��ԣ���ͬ���ϡ�����Ӿ�������Ƶ����ʵ��������룬����Ҫ�˽�ͬһ���ڲ�ͬϡ�����ӵIJ�ͬˮ����Ϊ�����۽��ͣ���ˣ�������ҪĿ����ȷ����ϡ����La��Ce��Pr��Nd�������ӵ�ˮ�Ͻṹ�����λ��(the coordination number�����CN)������ˮ��Һ�еĽṹ���������۵�ʵ���������۽ṹ����ϵ���ﵽ���Ӿ�ȷ�ؿ���ϡ�����ӽ����ͷ���[24]��ϡ�����ϵ��Ʊ����йػ�ѧ��Ӧ�Ľ��С�
1 �������
����Material studio(MS)������DMol3ģ�����ϡ��������ˮ���ﹹ���Ż������ù����ݶȽ���(GGA)�е�Perdew-Burke-Ernzerh(PBE)�����������������ӽ���������á�ɫɢ��������DFT-D��TS����������DSPP������ƣ�ȫ����(All electron)ԭ�Ӻ˴�����ʽ���۵��Ӳ�����DNPչ�������ǵ�ϡ��Ԫ�صĴ��ԣ������������������м��㾫������ΪFineˮƽ��������������λ������������Ϊ0.54 eV/nm��2.72��10-5 eV��5��10-4 nm���ضϰ뾶0.44 nm��
2 ������
2.1 �����Ż���Ĺ���
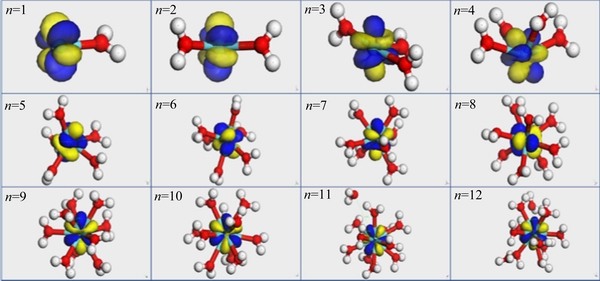
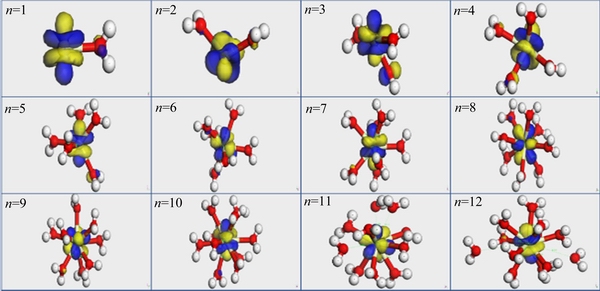
ˮ�е�H: O��O�ij���ԼΪ2.8 ����ʼģ�����õ�ϡ��������ˮ��������ԭ�ӵļ���(���Re��O����)Ϊ2.8 �������Ż���Re��O����С��2.8 �����ʾϡ��������ˮ��������������гɼ��Ŀ��ܣ���֮���ʾ������ų⡣����ϡ�������ڳɼ�ʱ5d�����4f����ijɼ�����[25-28]�������Ż���ɺ����������Զ����¼������й���ijɼ����(Monitor bonding)��ͼ1~4Ϊ�����Ż���ϡ������ˮ������ȶ����ͣ��Ը��ȶ����ͽ����˹���������㲢����LUMO���(Isosurface value=0.03a.u.)�����л�ɫ����Ϊ������Ϊ���ĵ����ƣ���ɫΪ������Ϊ���ĵ����ơ���1Ϊ�Ż���ĵļ��������ͼ5Ϊ�����Ż�������Ƶ�ϡ������ˮ�����Re��O�ļ���ͼ��
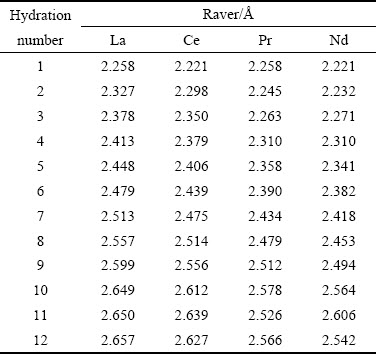
ͼ1~4��ʾ������ϡ�����Ӿ����Խ��1~10��ˮ���ӣ��γ�ˮ�Ͻṹ��(���¼��ˮ����)�������11��ˮ����ʱ��La��Ce��Pr��Nd��Χ�ֱ���1��3��2��1��ˮ�������룻�����12��ˮ����ʱ��La��Ce��Pr��Nd��Χ����2��ˮ�������롣����Ż�����ȶ����Ϳ�֪��La��Ce��Pr��Nd��һˮ���������ˮ����Ϊ10������λ��Ϊ10ʱ��ϡ����ˮ��������ԭ�ӵ�ƽ������ԼΪ2.6 �����ֳ������ԡ�
ͼ1 �����Ż���� La3+(H2O)n(n=1~12)
Fig. 1 Geometry optimized La3+(H2O)n(n=1-12)
ͼ2 �����Ż����Ce3+(H2O)n(n=1~12)
Fig. 2 Geometry optimized Ce3+(H2O)n(n=1-12)

ͼ3 �����Ż����Pr3+(H2O)n(n=1~12)
Fig. 3 Geometry optimized Pr3+(H2O)n(n=1-12)
ͼ4 �����Ż����Nd3+(H2O)n(n=1~12)
Fig. 4 Geometry optimized Nd3+(H2O)n(n=1-12)
��1 Re3+(H2O)n(n=1~12)��Re��Oƽ������
Table 1 Average bond length of Re��O in Re3+(H2O)n (n=1-12)
�����ϣ�ˮ�����ˮ����뾶R���㹫ʽ����
R=L(R��O)+L(O��H)*sin��
ʽ�У�L(R��O)Ϊ����ϡ��������ˮ������Oԭ�ӵļ�������L(O��H)Ϊˮ������Oԭ����Hԭ�ӵļ�����ԼΪ0.9 ����Ϊ��Զ��ˮ������Oԭ����Hԭ�������߶�l��ü���Oԭ�����ڵ㴦ˮ������ƽ����ļнǣ�(��)��
ͼ5 Re3+(H2O)n(n=1~12)��Re��O�ļ���
Fig. 5 Re��O bond length of Re3+(H2O)n(n=1-12)
Re3+��H2Oˮ���γ�ˮ������һ����̬ƽ����̣�L(R��O)��¾��DZ仯ֵ�����Ž��ˮ��������������Ϊ10ʱ��L(R��O)�����ȶ���ԼΪ2.6 ��ˮ���ӵļ���Ϊ104.5�㣬��̬ƽ����̣�����ԭ�Ӽ�ij�������Զ��ˮ���ӵ�Hԭ�Ӳ����������ƽ���֮�£����ǵ�ˮ���ӵĽṹ�ȶ��ԣ��µ�ȡֵ��ΧӦΪ37.75��~90�㡣��ˣ�����õ���ϡ������ˮ�����ˮ����뾶Ϊ3.22~3.59 ��
ϡ��Ԫ�س���6s��6p��5d�������ɼ�����������Ϊ9������ϡ����������λ����9���ҵ���Ҫԭ��[25-28]��ͬʱ����ͼ5��ʾ�������2~10��ˮ����ʱ��Re��O�����ij��Ⱦ�Ϊ��La��Ce��Pr��Nd�����Ӱ뾶ֵ��La3+(106 pm)��Ce3+(103 pm)��Pr3+(101 pm)��Nd3+(100 pm)�����ϡ��������ӵİ뾶Խ����ˮ���뾶Խ��һ�����[9, 29-31]��
���Ž��ˮ�������������ӣ�ϡ��ˮ��������ԭ�ӵ�ƽ��������������ȻС��2.8 ���ɴ˿ɼ����ڷ�Ӧ�Ĺ����У�ϡ������Һ�е�ˮ���Ӿ���һ�����������������⣬�ڶ���о�����[32-35]��ˮ�е�H: O��O�ij�����2.8 ���ң�ˮ���ӱ�����O: H��O�ij��ȴ�Լ��1.7 ���ң�����ϡ�����ӵ�Re��O������������֮�䣬˵����ˮ��Һ��ϡ����ˮ���ӵĽ����������ˮ����֮��Ľ��������
2.2 �����
����ϡ��������ˮ����֮���γ�ϡ��ˮ����ķ�Ӧ���£�
Re3++nH2O Re3+(H2O)n
Re3+(H2O)n
����������Ӧ����ʽ�������(EB)����ʽ����:
EB=EHydrates-ERe-nEH2O
Ϊ���о�����ˮ��������ת�����Ѷȣ��������·�Ӧʽ��������ˮ�������������ڽ����(EAB)��
Re3+(H2O)n+H2ORe3+(H2O)n+1
EAB=EB(n+1)-EB(n)-EH2O
����ϡ��������ˮ�Ľ���ܼ��������2��ʾ��
����ϡ��������ˮ��ϵĽ���ܾ�Ϊ����˵��ˮ������Ϊ���ȷ�Ӧ�����Ž��ˮ���ӵ���Ŀ���ӣ�Re3+��EBֵ��С����ˮ���ӵĽ���������������۽��ˮ����Ϊ���٣�����ܾ�Ϊ��La��Ce��Pr��Nd����֮ǰ�Աȼ��������Ӱ뾶�õ��Ľ������һ�¡�
ˮ�����Ŵ�������ļ�����0.1772~0.2022 eV (0.0078~0.0089 Ha)֮��[36]��OH��H2O�ļ���ԼΪ0.9088 eV (0.040 Ha)[37]������ϡ������EAB�ľ���ֵ����������ļ��ܣ�֤����ϡ����������ˮ���ʱ�γɼ����ȶ���Զ�����������n=1~9ʱ������ϡ�����ӵ�EAB�ľ���ֵ������OH��H2O�ļ��ܣ�ˮ���Ӹ�������ϡ�����ӽ�ϣ���n����9ʱ������ϡ�����ӵ�EAB�ľ���ֵ��ʼ����С��OH��H2O�ļ��ܵ��������ˮ��Һ�л����ϡ���������ǻ�����ˮ���ӵ���������뼸���Ż���ˮ�������ˮ�������������һ�¡�
2.3 ��ɷ���
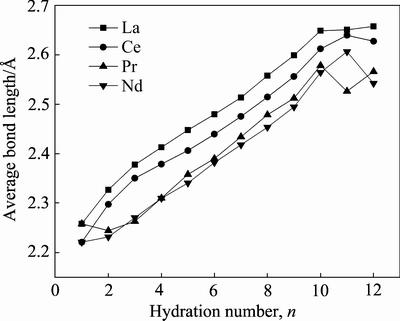
�ֱ���ESP������Mulliken������Re3+(H2O)n��������ϡ�������ĵ�ɷֲ������������3��ʾ��Re3+(H2O)n���γɹ�����һ�����ĵ��ת�ƹ��̡���ϵ���۽���������ˮ���Ĺ����д��ڵ��ӵ�ת�ƣ�������λ�������ӣ�Reԭ�ӴӸ���ˮ�����еõ����ӣ�Reԭ�ӵ�����ڼ��٣������Խ��ͣ�����ˮ������γɹ����е���ת�Ƶ�����ҪС�ö࣬ͨ��С��1e����������Re������ˮ��Һ����Ȼ�����������ԣ�����������+2e��+3e֮�䡣��nС��10ʱ������n�����ӣ�Re���ӵĵ������С������һˮ����������Re���ӵĵ�����������ȶ�����ʱ��ϡ������ˮ���ﻯѧ����Ҳ���ȶ���
��2 Re3+(H2O)n(n=1~12)�����EB��EAB
Table 2 Binding energy EB and EAB of Re3+(H2O)n(n=1-12)

��3 Re3+(H2O)n(n=1~12)��Re�ĵ�ɷֲ�
Table 3 Charge distribution of Re in Re3+(H2O)n(n=1-12)
2.4 ����Ӳ�Ⱥ�ǰ�߹������
Re3+��LewisӲ��[38-39]���зdz�ǿ�������ԣ���ˮ���ӵĽ�Ϸ�����Ӳ��������С�Ӳ��Ӳ���Ľ��ԭ��[40]��LI��YAN��EVANS[41]֤����ǰ�߹�����ۿ�������Ϊ��Ӳ���(HSAB)ԭ����һ���֡�Ӳ����������е������Ŀյļ۹������ LUMO�����Ӳ��������Ǿ��п��ṩ�ĵ������ļ۲�HOMO���Ӷ�[40, 42]��
�����Ż���ϡ������ˮ�����LUMO���(Isosurface value=0.03a.u.)��ʾ��ͼ1~4�С��۲�ϡ��ˮ�����LUMO��������Ϳ�֪������ˮ�������������ӣ�ϡ��������Χ��LUMO�����״���ֳ������Ա仯������La3+(H2O)nΪ������n�ֱ����1��2��4��5��6��7��9��10��11��12ʱ��LUMO������������ơ���Ϊ��ѧ��Ӧ����͵������չ����LUMO����ĸı�˵����ϵ��������ĸı䣬��һ��˵���ڽ��ˮ�Ĺ����з����˵��ӵ�ת�ơ������9��ˮ�����Ժ�����ϡ�����ӵ�Re��O��������Ȼ��������ı仯���ҿ����ڵ�һˮ�����ڶ�ˮ����������ˮ���ӣ���LUMO�������״�仯��С��˵����ʱ����ϵ�Ѿ������ȶ�����֮���Ӧ�ģ����Ž��ˮ�����ӣ�ϡ��ˮ�����LUMOֵ��������ٶ��Ż�����˵��ϡ��Ԫ�ص�ˮ�������͡�����ϡ������ˮ�����LUMO��HOMO����������4��ʾ��
ʹ�����ʵ�LUMO��HOMO������������������ı�ʾ���ʵ�Ӳ�ȡ���ˣ����IJ����������ַ�ʽ��ʾˮ�����Ӳ�ȣ�
1) ʹ��ˮ����������LUMO�������ֵ��HOMO�������ֵ�IJ�����ʾˮ����������Ӳ�ȣ�
��E=ELUMO-EHOMO
������Խ����Ӳ��Խ��������Ӳͨ������γɵ���λ���й��ۼ��Ĺ���������[43]��
2) ʹ��ˮ�����LUMO�������ֵ��ˮ���ӵ�HOMO�������ֵ�IJ�ľ���ֵ����ʾˮ���������ˮ���ӵ�Ӳ�ȣ�
|��E|=EHydrates/LUMO-EH2O/HOMO
����ϡ������ˮ�����|��E|���E���������5��ʾ��
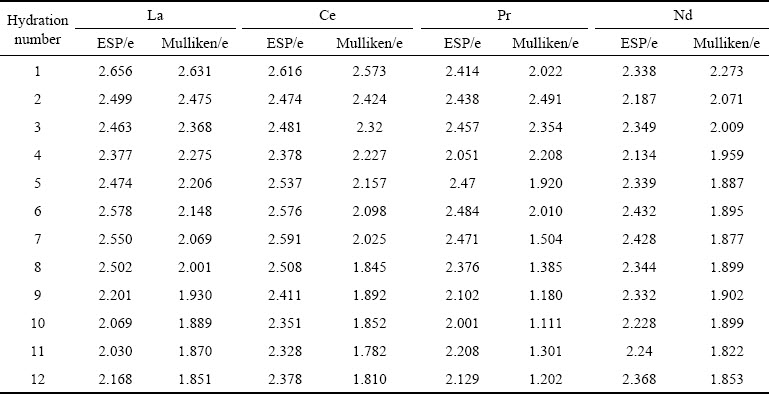
��4 Re3+(H2O)n(n=1~12)��Re��HOMO�����LUMO�������
Table 4 Energy calculation results of HOMO orbital and LUMO orbital of Re in Re3+(H2O)n(n=1-12)
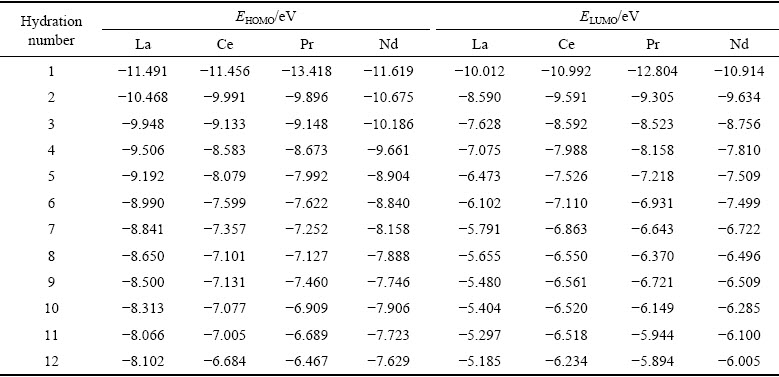
��5 Re3+(H2O)n(n=1~12)��Re��|��E|���E������
Table 5 Calculation results of |��E| and ��E of Re in Re3+(H2O)n(n=1-12)
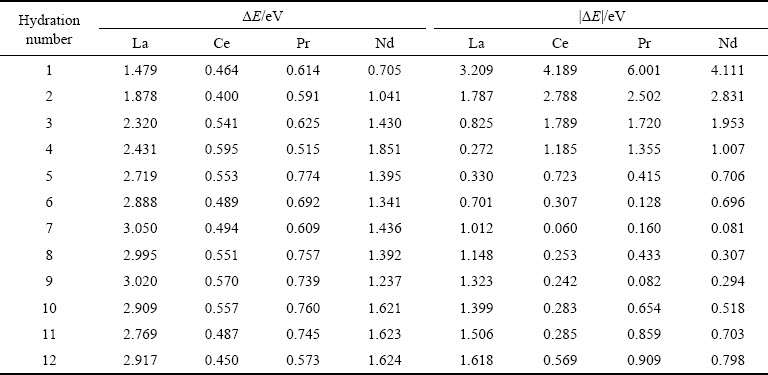
��ͼ6��ʾ������ϡ������������Ӳ��˳��Ϊ��La��Nd��Pr��Ce��ϡ��Ԫ��ʧȥ���ӵ�˳��Ϊ:�����6s���ӣ������5d���ӣ�����������4f���ӡ����γ����۽���������ʱ��ϡ��Ԫ�ص������������з����仯�����۵ĺ�������Ų�Ϊ��La3+:[Xe]��Ce3+:[Xe]4f1��Pr3+:[Xe]4f2��Nd3+:[Xe]4f3����ʱ��La3+������HOMO���Ϊ5p�����Ce3+��Pr3+��Nd3+������HOMO���Ϊ4f�����La3+��Ce3+������LUMO���Ϊ5d�����Pr3+��Nd3+������LUMO���Ϊ6s���������Pauling�����ܼ�ͼ��Cottonԭ�ӹ���ܼ�ͼ��֪������La3+��HOMO��LUMO�����Խ�������ܼ��飬��E���Ӳ����ߣ�Ce3+��Pr3+��Nd3+��HOMO��LUMO���ͬ��һ���ܼ���֮�ڣ���Eֵ�IJ����Ҫ�ܺ��������Ӱ�졣
��ͼ7��ʾ���ֱ�Ϊϡ������ˮ�����LUMOֵ��ˮ��HOMO�IJ�ֵ�������ֵ���ڽ��ˮ�Ĺ����У���ϵ�ġ�Eֵ���La��Ce��Pr��Nd�ֱ���4��6��7��7��ˮʱ����ϵ�ĵ�|��E|���0��˵����ˮ��Һ�����У�Re3+������������ˮ��ˮ����Ϊ4��6��7��7ʱ������H2O����ˮ�ϡ��ӽ���ܵĴ�С����������ϵ�H2O����һ��ʱ������La��H2O�Ľ������ǿ������ʱ��La���ܽ�ϵ���С����H2O��ȴ���٣�Ce��Pr��Nd���ܽ�ϵ���С����H2O�����ơ����ӹ��������Ϊ����ֵ֮��ľ���ֵ(|��E|)ԽС������Ӧ��֮�������þ�Խǿ����ͬ�Ļ����£�ˮ�����ṩ��HUMO���������ͬ��������ϡ�����Ӿ��в�ͬ��LUMO������La��LUMO����ֵ��ߣ�Ce��Pr��Nd��LUMO����ֵ�����ҵ���La�����ԣ�ͬΪ��ϡ���������ϡ�����ӱ��ֳ���ͬ����С���ܽ�ϵ�ˮ��������
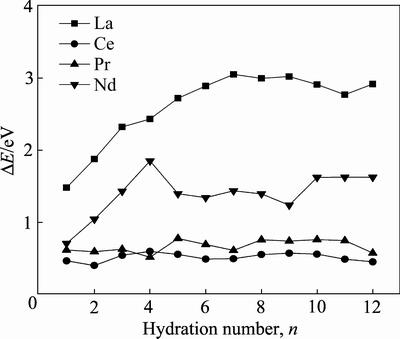
ͼ6 Re3+(H2O)n(n=1~12)��Re���ӡ�E�仯
Fig. 6 Re ion ��E in Re3+(H2O) n(n=1-12)��
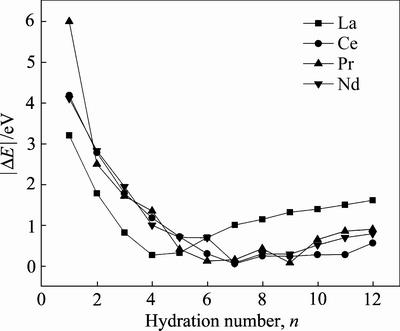
ͼ7 Re3+(H2O)n(n=1~12)��Re����|��E|�仯
Fig.7 Re ion |��E| in Re3+(H2O)n(n=1-12)
2.5 ����
��PBE����������Re3+(H2O)n�صĺ������չ��ס�������OH��������IJ������㣬���������ΪRe3+(H2O)n���о��ṩ��ϸ����Ϣ��ͼ8��ʾΪLa3+(H2O)n(n=1~12)�صĺ�����ס�La3+(H2O)n�ĶԳ�������(Vsym)��λ��Լ3546��3620��3640��3671��3687��3695��3698��3704��3704��3708��3708��3698 cm-1�������Գ�������(Vasym)��λ��Լ3629��3686��3733��3754��3764��3780��3789��3792��3791��3791��3807��3804 cm-1�������Կ�����n=1~6ʱ������n�����ӣ�Vsym��Vasym��������ƣ��������ӵ���ܼ����ͼ��ٴӽ������ӵ�ˮ���ӵĵ��ת����һ�µģ� Zn2+(H2O)n[44]��Cd2+(H2O)n[44]��Hg2+(H2O)n[44]��Ni+(H2O)n[45]��K+(H2O)n[46]��Ca2+(H2O)n[47]ˮ����Ҳ�۲쵽���������ơ����ǣ���ͼ8��ʾ����n=7~10ʱ��Vsym��Vasym���������С���ر�ģ���n=11��12ʱ��Vsym��Vasym������������ƣ������ɷ������һ�¡�
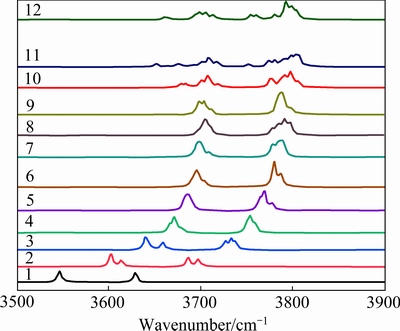
ͼ8 ģ��ṹ��La3+(H2O)n(n=1~12)�ĺ������
Fig. 8 Infrared spectrum of La3+(H2O)n(n=1-12) with simulated structure
3 ����
1) ��ϡ������La3+��Ce3+�� Pr3+��Nd3+�ܹ���ˮ��Һ���γ��ȶ��ĵ�һˮ���㣬ǰ�߹�����������ʾ���ȶ���ˮ����n��С��4��6��7��7����һˮ������������10��ˮ���ӡ�
2) �γ��ȶ��ĵ�һˮ�������ϡ������La3+��Ce3+��Pr3+��Nd3+��ˮ����Re3+(H2O)n�����ʳ��ֳ������Ժ������ԡ���ͬˮ���������£�Re��Oƽ��������La��Ce��Pr��Nd��Re3+(H2O)10��Re��Oƽ������ԼΪ2.6 ����ϡ������ˮ�����ˮ����뾶ԼΪ3.22~3.59 ����ϡ�����ӵ�ˮ����Ӧ��Ϊ���ȷ�Ӧ����ˮ������λ����ͬʱ������ܴ�С˳��Ϊ��La��Ce��Pr��Nd����ɷ��������ʾ��Re���ӵĵ��������2e���ҡ�ǰ�߹�����������ʾ������ϡ������������Ӳ��˳��Ϊ��La��Nd��Pr��Ce��
3) ��һˮ�������ˮ��������ϡ������La3+��Ce3+�� Pr3+��Nd3+�Ľ��ǿ���������ڵ�һˮ�����ڵ�ˮ���ӡ���n����10ʱ��Re��O�������Ը���������ܱ仯��С��Re���ӵĵ�����仯���Լ�С��ǰ�߹������ֵ�仯��С�� La3+(H2O)n�ĺ������Vsym��Vasym�岻�ڷ������ơ�
REFERENCES
[1] LIM X. Chemistry: Degrees of separation[J]. Nature, 2015, 520(7548): 426-427.
[2] TEYCHENE J, BALMANN H R, MARON L, et al. Investigation of ions hydration using molecular modeling[J]. Journal of Molecular Liquids, 2019, 294: 111394.
[3] ABDELSALAM H, TELEB N H, YAHIA I S, et al. First principles study of the adsorption of hydrated heavy metals on graphene quantum dots[J]. Journal of Physics and Chemistry of Solids, 2019, 130: 32-40.
[4] CLAVAGUERA C, POLLET R, SOUDAN J M, et al. Molecular dynamics study of the hydration of lanthanum(��) and europium(��) including many-body effects[J]. The Senior Engineer Journal of Physical Chemistry B, 2005, 109(16): 7614-7616.
[5] SESSA F, SPEZIA R, D'ANGELO P. Lutetium(��) aqua ion: On the dynamical structure of the heaviest lanthanoid hydration complex[J]. Journal of Chemical Physics, 2016, 144(20).
[6] TIRLER A O, PASSLER P P, RODE B M. The lanthanoid hydration properties beyond the ��Gadolinium Break��: Dysprosium(��) and holmium(��), an ab initio quantum mechanical molecular dynamics study[J]. Chemical Physics Letters, 2015, 635: 120-126.
[7] MARJOLIN A, GOURLAOUEN C, CLAVAGUERA C, et al. Hydration Gibbs free energies of open and closed shell trivalent lanthanide and actinide cations from polarizable molecular dynamics[J]. Journal of Molecular Modeling, 2014, 20(10).
[8] ZHANG J, HEINZ N, DOLG M. Understanding Lanthanoid(��) Hydration Structure and Kinetics by Insights from Energies and Wave functions[J]. Inorganic Chemistry, 2014, 53(14): 7700-7708.
[9] HOFER T S, WEISS A K H, RANDOLF B R, et al. Hydration of highly charged ions[J]. Chemical Physics Letters, 2011, 512(4): 139-145.
[10] FUJIWARA T, MORI H, MOCHIZUKI Y, et al. Theoretical study of hydration models of trivalent rare-earth ions using model core potentials[J]. Journal of Molecular Structure: Theochem, 2010, 949(1): 28-35.
[11] BUZKO V, SUKHNO I, BUZKO M. Ab initio and DFT study of Lu3+ hydration[J]. Journal of Molecular Structure: THEOCHEM, 2009, 894(1): 75-79.
[12] SMIRNOV P, KRITSKII I, GRECHIN O. Models of the nearest surrounding of ions in aqueous solutions of dysprosium chloride[J]. Russian Journal of Physical Chemistry A, 2016, 90(2): 406-410.
[13] HABENSCHUSS A, SPEDDING F H. The coordination (hydration) of rare earth ions in aqueous chloride solutions from X-ray diffraction. ��. TbCl3, DyCl3, ErCl3, TmCl3 and LuCl3[J]. The Journal of Chemical Physics, 2008, 70(6): 2797.
[14] HABENSCHUSS A, SPEDDING F H. The coordination (hydration) of rare earth ions in aqueous chloride solutions from X-ray diffraction. ��. LaCl3, PrCl3 and NdCl3[J]. The Journal of Chemical Physics, 2008, 70(8): 3758.
[15] HABENSCHUSS A, SPEDDING F H. The coordination (hydration) of rare earth ions in aqueous chloride solutions from X-ray diffraction. ��. SmCl3, EuCl3 and series behavior[J]. The Journal of Chemical Physics, 2008, 73(1): 442.
[16] NASLUND J, LINDQVIST-REIS P, PERSSON I, et al. Steric effects control the structure of the solvated lanthanum(��) ion in aqueous, dimethyl sulfoxide, and N,N��-dimethylpropyleneurea solution. An EXAFS and large-angle X-ray scattering study[J]. Inorganic Chemistry, 2000, 39(18): 4006-4011.
[17] D'ANGELO P, ZITOLO A, MIGLIORATI V, et al. Revised ionic radii of lanthanoid(��) ions in aqueous solution[J]. Inorganic Chemistry, 2011, 50(10): 4572-4579.
[18] PERSSON I, D'ANGELO P, De PANFILIS S, et al. Hydration of lanthanoid(��) ions in aqueous solution and crystalline hydrates studied by EXAFS spectroscopy and crystallography: The myth of the ��gadolinium break��[J]. Chemistry-A European Journal, 2008, 14(10): 3056-3066.
[19] D'ANGELO P, ZITOLO A, MIGLIORATI V, et al. Analysis of the detailed configuration of hydrated lanthanoid(��) ions in aqueous solution and crystalline salts by using K- and L3-Edge XANES spectroscopy[J]. Chemistry-A European Journal, 2010, 16(2): 684-692.
[20] RUDOLPH W, IRMER G. On the hydration of heavy rare earth ions: Ho3+, Er3+, Tm3+, Yb3+ and Lu3+: A Raman study[J]. MOLECULES, 2019, 24(10).
[21] RUDOLPH W W, IRMER G. Raman spectroscopic characterization of light rare earth ions: La3+, Ce3+, Pr3+, Nd3+ and Sm3+ hydration and ion pair formation[J]. Dalton Transactions, 2017, 46(13): 4235-4244.
[22] RUDOLPH W, IRMER G. Hydration and ion pair formation in aqueous Lu3+ solution[J]. Molecules, 2018, 23(12): 3237.
[23] MARTELLI F, ABADIE S, SIMONIN J, et al. Lanthanoids(��) and actinoids(��) in water: Diffusion coefficients and hydration enthalpies from polarizable molecular dynamics simulations[J]. Pure and Applied Chemistry, 2012, 85(1): 237-246.
[24] YU D, DU R, XIAO J, et al. Theoretical Study of pKa values for trivalent rare-earth metal cations in aqueous solution[J]. The Journal of Physical Chemistry A, 2018, 122(2): 700-707.
[25] ��ӽ��, ���ҳ�. ϡ���Ľṹ�ص���4f����ڳɼ��е�����[J]. ϡ��, 2008(2): 100-101.
LI Yong-feng, KUANG Ju-chi. Structure feature of RE and effect of its 4f orbital in bonding process[J]. Chinese Rare Earths, 2008(2): 100-101.
[26] ����. ϡ��4f5d���ӳɼ����Լ����ڵ�������нṹ�����ʵĵ�г[D]. �Ϸ�: �й���ѧ������ѧ, 2019.
ZHANG Li-fang. Rare-earth 4f/5d electrons and their bonding characters in modulation both on structure and properties of conductive materials[D]. Hefei: University of Science and Technology of China, 2019.
[27] �� ·. ����ϡ������4f-4f ��4f-5dԾǨ�Ĺ�ѧ�¶ȴ���[D]. �Ϸ�:�й���ѧ������ѧ, 2019.
ZHAO Lu. Optical temperature sensing based on 4f-4f and 4f-5d transitions of rare earth ions[D]. Hefei: University of Science and Technology of China, 2019.
[28] �� ο. ��ϵԪ��4f�����+2/+3�ۻ������еijɼ�����[D]. �Ϻ�: �Ϻ���ͨ��ѧ, 2015.
XU Wei. The bonding properties of lanthanide 4f orbits in di-/trivalent compounds[D]. Shanghai: Shanghai Jiao Tong University, 2015.
[29] SUN C Q, HUANG Y, ZHANG X. Hydration of Hofmeister ions[J]. Advances in Colloid and Interface Science, 2019, 268: 1-24.
[30] SOLDATOV V, ZELENKOVSKII V, KOSANDROVICH E. Hydration of ion exchangers: thermodynamics and quantum chemistry calculations. ��. An improved variant of the predominant hydrates model[J]. Reactive and Functional Polymers, 2016, 102: 147-155.
[31] SOLDATOV V, ZELENKOVSKII V, KOSANDROVICH E. Hydration of ion exchangers: Thermodynamics and quantum chemistry calculations. ��. The state of the proton and water molecules in hydrogen form of sulfostyrene ion exchangers[J]. Reactive and Functional Polymers, 2016, 102: 156-164.
[32] SIGALA P A, RUBEN E A, LIU C W, et al. Determination of hydrogen bond structure in water versus aprotic environments to test the relationship between length and stability[J]. Journal of the American Chemical Society, 2015, 137(17): 5730-5740.
[33] ZNAMENSKIY V S, GREEN M E. Quantum calculations on hydrogen bonds in certain water clusters show cooperative effects[J]. Journal of Chemical Theory and Computation, 2007, 3(1): 103-114.
[34] MOLLER K B, REY R, HYNES J T. Hydrogen bond dynamics in water and ultrafast infrared spectroscopy: A theoretical study[J]. The Journal of Physical Chemistry A, 2004, 108(7): 1275-1289.
[35] UMEYAMA H, MOROKUMA K. The origin of hydrogen bonding. An energy decomposition study[J]. Journal of the American Chemical Society, 1977, 99(5): 1316-1332.
[36] WENDLER K, THAR J, ZAHN S, et al. Estimating the hydrogen bond energy[J]. The Journal of Physical Chemistry A, 2010, 114(35): 9529-9536.
[37] MELTON C E. Formation of, by termolecular reactions, and bond dissociation energy, structure and bond length for OH-H2O and O-H2O[J]. The Journal of Physical Chemistry, 1972, 76(22): 3116-3120.
[38] ������. �����Ӳ�ȵļ��������[J]. ��ѧͨ��, 1976(6): 26-32.
LIU Qi-tao. Key parameter scale of acid-base softness and hardness[J]. Chemistry, 1976(6): 26-32.
[39] �� ��, �� �t, �� ��, ��. ������Ӳ������۵ĵ���ɢ����ϡ�������������װ�Ŀɿغϳ�[J]. ��ѧѧ��, 2013, 71(3): 360-366.
GU Jun, DING Yi, KE Jun et al. Controllable synthesis of monodispersed middle and heavy rare earth oxysulfide nanoplates based on the principles of HSAB theory[J]. Acta Chimica Sinica, 2013, 71(3): 360-366.
[40] ����Ӣ, ��³��, ��СӢ. ��Ӳ�������Ӧ��[J]. ��ͷ��ѧҽѧԺѧ��, 2000(2): 17-19.
SHEN Wen-ying, ZHANG Lu-mian, HE Xiao-ying. Application of hard and soft acids and bases rule[J]. Journal of Shantou University Medical College, 2000(2): 17-19.
[41] LI Y, EVANS J N S. The Fukui function: A key concept linking frontier molecular orbital theory and the hard-soft-acid-base principle[J]. Journal of the American Chemical Society, 1995, 117(29): 7756-7759.
[42] ������, ӡ����. �����Ӳ�ȵ��Ʊ���ڸ�ѡ���ṹ�����о���Ӧ��[J]. ������ѧѧ��(��Ȼ��ѧ��), 2013, 34(7): 1035-1038.
WANG Ji-zhen, YIN Wan-zhong. Application of acid-base potential scale in structure performance study of flotation reagents[J]. Journal of Northeastern University(Natural Science), 2013, 34(7): 1035-1038.
[43] PEARSON R G. Recent advances in the concept of hard and soft acids and bases[J]. Journal of Chemical Education, 1987, 64(7): 561.
[44] YUAN X, ZHANG C. Density functional theory study on the inner shell of hydrated M2+(H2O)1-7 cluster ions for M=Zn, Cd and Hg[J]. Computational and Theoretical Chemistry, 2019: 112666.
[45] WALTERS R S, PILLAI E D, DUNCAN M A. Solvation dynamics in Ni+(H2O)n clusters probed with infrared spectroscopy[J]. Journal of the American Chemical Society, 2005, 127(47): 16599-16610.
[46] ZHU F, ZHOU H, ZHOU Y, et al. The investigation of structure and IR spectra for hydrated potassium ion clusters K+(H2O)n=1-16 by density functional theory[J]. The European Physical Journal D, 2016, 70(11).
[47] BUSH M F, SAYKALLY R J, WILLIAMS E R. Hydration of the calcium dication: Direct evidence for second shell formation from infrared spectroscopy[J]. Chem Phys Chem, 2007, 8(15): 2245-2253.
DFT study on light rare earth ion hydrate Re3+(H2O)n (n=1-12, Re = La, Ce, Pr, Nd)
OU Jia-cai1, 2, ZHANG Tian-xi1, 3, HUANG Li-jin-hong4, WU Bo-zeng5, HUANG Wan-fu1
(1. School of Resource and Environmental Engineering, Jiangxi University of Science and Technology. Ganzhou 341000, China;
2. Guangxi Wuxin Mining Investment, Subsidiary of China Minmetals Rare Earth Group Co., Ltd., Nanning 530022, China;
3. WeifangLongda Zinc Industry Co. LTD., Weifang 262100, China;
4. School of Architecture and Design, Jiangxi University of Science and Technology, Ganzhou 341000, China;
5. School of Chemical and Environmental Engineering, China University of Mining & Technology, Beijing 100083, China)
Abstract: DMol3 module of Material Studio was used to study the hydrate Re3+(H2O)n(n=1-12) of four light rare earth ion coordination water quantities of La, Ce, Pr and Nd. The geometric structure, binding energy, frontier molecular orbital, charge and vibration of the geometrically optimized configuration were analyzed. The results show that the minimum hydration number of the first hydration layer of La, Ce, Pr and Nd are 4, 6, 7 and 7 respectively, which can contain up to 10 water molecules. The average Re��o bond length of La��Ce��Pr��Nd, the average Re��O bond length of Re3+(H2O)10 was about 2.6 , the hydration layer width of light rare earth ionic hydrate was about 3.22-3.59 . The hydration reaction of light rare earth ions is exothermic. When the coordination number of water molecules is the same, the order of binding energy is La��Ce��Pr��Nd. Charge analysis results show that rare earth ions gain electrons in the hydration reaction. When the first hydration layer is filled, the properties of Re3+(H2O)n gradually become stable, and the charge of Re ions is around 2e. The results of frontier molecular orbital analysis show that, different from the change of ion radius, the hardness sequence of the four rare earth ions is La��Nd��Pr��Ce. The infrared spectrum of La3+(H2O)n was simulated. With the increase of the number of water molecules, the Vsym and Vasym peaks showed blue shift. When n was greater than 10, the Vsym and Vasym peaks did not show blue shift, indicating that the water molecules outside the first hydration layer did not react with La ions.
Key words: rare earth; ionic hydrate; density functional; Material studio
Foundation item: Project(41362003) supported by the National Natural Science Foundation of China
Received date: 2020-12-03; Accepted date: 2021-08-03
Corresponding author: HUANG Wan-fu; Tel: ; E-mail: Sim2008@sina.com
(�༭ )
������Ŀ��������Ȼ��ѧ����������Ŀ(41362003)
�ո����ڣ�2020-12-03�������ڣ�2021-08-03
ͨ�����ߣ��������ڣ���ʿ���绰�� ��E-mail��Sim2008@sina.com

