镁合金阳极氧化的研究进展与展望
来源期刊:中国有色金属学报2006年第7期
论文作者:张荣发 单大勇 韩恩厚 曾志良
文章页码:1136 - 1148
关键词:镁合金; 腐蚀与防护; 阳极氧化; 电解液; 机制
Key words:magnesium alloys; corrosion and protection; anodization; electrolyte; mechanism
摘 要:回顾镁合金阳极氧化历史, 介绍制备工艺、 电解液组成及作用, 同时对镁合金阳极氧化机理进行探讨。 随着人类环保意识的增强, 世界能源的紧缺和氧化设备的不断更新, 认为电参数如频率、 占空比、 电压和电流密度对氧化膜性能的影响、 阳极氧化电流效率的测定、 氧化膜扩散规律的研究和环保型电解液的开发为未来镁合金阳极氧化研究的重点。
Abstract: The history of anodic oxidization on magnesium and its alloys was simply reviewed, and its treating process, compositions and functions of electrolyte were introduced comprehensively. In addition, the mechanism of oxidization on magnesium was discussed. With the development of human awareness on environmental protection and the scarcity of world resources and upgrade of anodization equipment, the effects of electric parameters on properties of anodic coating, measurement of current efficiency, study of diffusion regularity for anodic coating formation and development of environmentally friendly electrolyte were put forward about anodization.
文章编号: 1004-0609(2006)07-1136-13
张荣发1, 2, 单大勇2, 韩恩厚2, 曾志良3
(1. 江西科技师范学院 江西省材料表面工程重点实验室, 南昌 330013;
2. 中国科学院 金属研究所 材料环境腐蚀试验研究中心, 沈阳 110016;
3. 江西科技师范学院 理工学院, 南昌 330013)
摘 要: 回顾镁合金阳极氧化历史, 介绍制备工艺、 电解液组成及作用, 同时对镁合金阳极氧化机理进行探讨。 随着人类环保意识的增强, 世界能源的紧缺和氧化设备的不断更新, 认为电参数如频率、 占空比、 电压和电流密度对氧化膜性能的影响、 阳极氧化电流效率的测定、 氧化膜扩散规律的研究和环保型电解液的开发为未来镁合金阳极氧化研究的重点。
关键词: 镁合金; 腐蚀与防护; 阳极氧化; 电解液; 机制
中图分类号: TG174 文献标识码: A
ZHANG Rong-fa1, 2, SHAN Da-yong2, HAN En-hou2, ZENG Zhi-liang3
(1. Jiangxi Key Laboratory of Surface Engineering,
Jiangxi Science and Technology Normal University, Nanchang 330013, China;
2. Environmental Corrosion Center, Institute of Metal Research,
Chinese Academy of Sciences, Shenyang 110016, China;
3. Institute of Technology, Jiangxi Science and Technology Normal University,
Nanchang 330013, China)
Abstract: The history of anodic oxidization on magnesium and its alloys was simply reviewed, and its treating process, compositions and functions of electrolyte were introduced comprehensively. In addition, the mechanism of oxidization on magnesium was discussed. With the development of human awareness on environmental protection and the scarcity of world resources and upgrade of anodization equipment, the effects of electric parameters on pro-perties of anodic coating, measurement of current efficiency, study of diffusion regularity for anodic coating formation and development of environmentally friendly electrolyte were put forward about anodization.
Key words: magnesium alloys; corrosion and protection; anodization; electrolyte; mechanism
镁合金具有密度小, 比强度、 比刚度高, 电磁屏蔽性、 减震性好以及优良的切削加工和抛光性能, 在航空、 汽车和3C产品领域具有很大的应用潜力[1-4], 但是其耐蚀性较差, 这使镁合金的应用受到限制。 提高镁合金耐蚀性的方法有许多种, 有人对此进行了综述[5-7]。 普遍认为阳极氧化是使镁和镁合金获得最佳耐蚀性的方法[8], 另外镁合金阳极氧化膜还具有与基体金属结合力强、 电绝缘性好、 光学性能优良及耐磨损等优点, 同时具有多孔结构, 能够按照要求进行着色/封孔处理, 并能为进一步涂覆有机涂层如油漆等提供优良底层, 是一种很有前途的表面处理技术。 阳极氧化技术产生于20世纪20年代[9], 直到1951年以后, HAE和DOW17工艺的相继出现才使阳极氧化技术在镁合金防护处理中应用成为可能。 随着人们的不断探索, 镁阳极氧化发展趋势之一是不断升高工作电压。 1955年前后出现的Cr-22工艺, 其工作电压最高可达380V[10]。 1994年, Zozulin等[11]对镁的高电压(高达340V)阳极氧化行为进行了较为深入的研究, 并获得较DOW17和HAE工艺所得膜层耐蚀性更佳的膜层。
随着阳极氧化工作电压的不断提高, 有人根据由工作电压不同造成阳极氧化现象及所得膜层性能不同, 将阳极氧化电压―电流曲线划分为法拉第区、 火花放电区和弧光放电区3个区间[12](图1), 并将对应于火花放电区中电压较高区域的阳极氧化称为微弧氧化(MAO)[13-15]、 等离子体溶液氧化(PEO)[16] 、 等离子体阳极氧化(Plasma anodizing)[17] 或阳极火花沉积(Anodic spark deposition, ASD)[18]。
微弧氧化的研究可以追溯到1932年, 由两个德国科学家Gunterschulze 和Betz首先开始进行, 然后美国法兰克福兵工厂的研究人员和伊利诺斯大学的教授以及前东德的人员为该项技术的发展做出了贡献。 俄国科学家Markov被称为这项技术之父, 由于他于20世纪70年代成功地将此项技术从实验室研究转向工业应用[16-18]。
1 镁合金阳极氧化工艺
阳极氧化工艺一般包括前处理、 阳极氧化和后处理工艺。
1.1 前处理
试样在阳极氧化之前, 必须进行前处理。 前处理是诸如电镀、 化学镀、 化学转化膜等表面技术中极为关键的步骤。 镁合金阳极氧化之前, 一般都须用水砂纸由粗到细依次打磨, 用水洗净后再用丙酮脱脂。
镁合金阳极氧化前处理方法还包括热碱洗, 它的目的在于清除金属表面的油脂和其它有机污染物。 当镁(合金)表面有氧化物、 腐蚀产物等存在时, 可在碱洗之后作进一步的酸洗处理, 所用的酸一般为稀硫酸、 稀硝酸或铬酐溶液等。 专利[19, 20]所用的碱洗溶液为NaOH 5g/L, Na3PO4 1g/L, 合成肥皂1g/L; 酸洗溶液为H3PO4(85%) 380mL/L, H2SO4(98%) 16mL/L, H2O 604mL/L。
在碱洗过程中, 试样不会有质量损失, 有时质量甚至会增加; 使用110mL/L HNO3+120g/L CrO3的酸洗溶液, 质量损失很大, 当工件尺寸要求很严格时, 不能使用[21]。
另外, 一些专利还采用了在氟化物中活化这种前处理工艺, 氟化物可以是氢氟酸和NH4F, 也可以是NH4HF2。 这是由于镁合金中α相和β相(Mg17Al12)的腐蚀电位分别接近-1.73V(0.1mol/L Calomel electrode)和-1.0V (0.1mol/L Calomel electrode)。 在氟化物中进行活化处理, 可形成极难溶的MgF2沉淀在样品表面, 使样品表面电位尽量相等, 避免局部电偶腐蚀的发生[22]。 典型的表面前处理方法如表1所列[23]。
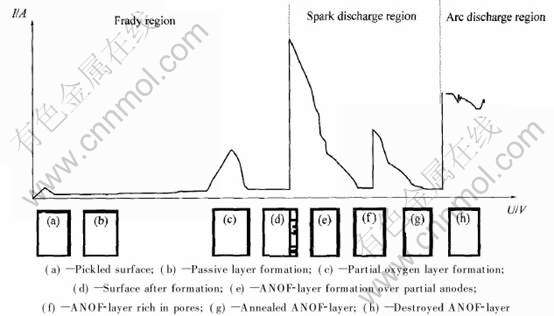
图1 阳极火花沉积模型
Fig.1 Model of anodic oxidation by spark discharge (ANOF)
1.2 阳极氧化
对镁合金阳极氧化时, 镁作为阳极, 不锈钢、 铁、 镍或导电性电解池本身为阴极。 水溶液电解伴随着许多过程。 阳极表面既可能析出氧气也可能是镁的氧化, 阴极表面既可能析出氢气也可能是阳离子的还原(见图2)[16]。
电解槽通电后, 阴离子向阳极移动, 阳离子向阴极移动, 当电压达到一定值时, 阳极上形成氧化[CM(22]膜。 且随着氧化时间的延长, 膜的厚度不断增加, [CM)] 外加电压也增大。 当外加电压大于膜的击穿电压时, 膜被击穿, 在试样上可以观察到火花产生, 同时伴随着气体析出, 这时电压表指针不断摆动。
击穿电压仅与电解液的组成、 浓度和被氧化的金属基体有关, 浓度越大, 击穿电压越低[24]。
击穿电压与电解质电阻的经验公式为[25]
![]()
式中 aB、 bB对于给定的金属和电解液组成为常数;
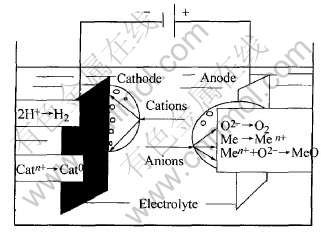
图2 电解质水溶液中的电极过程
Fig.2 Electrode processes in electrolysis of aqueous solution
ρ为电解质电阻。
产生火花时, 一方面可以使膜快速生长, 同时使膜孔隙增大, 对膜的耐蚀性不利; 另一方面, 在通常的镁阳极氧化过程中, 等离子放电的火花发生位置在工件表面70nm之内, 这种局部高热能冲击可能对工件的力学性能不利[26]。 阳极氧化装置还包括搅拌冷却设备、 搅拌电解液以及低的电解液温度, 可以使氧化物/电解液表面更好地冷却, 因而膜的孔隙更少, 并且形貌更均匀[27]; 搅拌的另一个效果是抑制副反应, 尤其是气体在两个电极上的析出[28]。
1.3 后处理
普通阳极氧化处于法拉第区, 所得膜层呈多孔结构; 微弧氧化处于火花放电区中电压较高的区域, 所得膜层均匀, 孔隙的相对面积较小[29]。 普通阳极氧化膜如果不进行后处理, 盐雾实验一般不超过100h; 而微弧氧化膜可以不封孔, 为了提高耐蚀性可采用涂漆的方法。
镁合金阳极氧化膜封孔试剂有多种, 铬酸盐溶液封孔用得较早, 效果也好, 如HAE就用它封孔。 由于六价铬毒性大且致癌, 因此现在很少采用。 专利中用得较多的是硅酸盐封孔[19, 20], 要求阳极氧化后的镁工件先在硅酸钠水溶液中加热15min, 然后将试样放入空气中30min, 空气中的CO2会与试样上残留的水玻璃发生反应, 生成SiO2, 从而封住孔隙。 反应式为
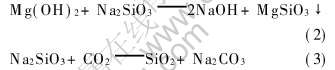
蒋百灵等[30]将在磷酸盐与硅酸盐复合体系电解液中微弧氧化处理的样品用石蜡、 丙烯酸和沸水溶液封孔, 于10% NaCl盐雾中进行腐蚀实验, 结果表明: 石蜡封孔耐蚀性效果最好, 其机理是由于[CM(22]融化的石蜡被吸附到膜层表面, 充入微孔, 使受腐[CM)]
表1 表面预处理方法[23]
Table 1 Surface pre-treatment methods[23]

蚀的有效接触面积大大减小, 提高了膜层的耐蚀性。
采用在碱金属的磷酸二氢盐溶液中封孔也有报道[31, 32]。 常见的后处理方法见表2。
表3列出了部分专利和文献介绍的前处理、 后处理工艺及电解液组成。
表2 阳极氧化工艺的后处理方法[23]
Table 2 Post-treatment of anodizing process[23]

表3 部分镁及镁合金阳极氧化工艺
Table 3 Some anodizing processes of magnesium and its alloys

2 电解质
影响镁合金阳极氧化成膜效果的因素包括电解液的组成及其浓度、 电参数(电压、 电流)类型、 幅值及其控制方式、 溶液温度、 pH值及处理时间等, 其中电解液组分是镁阳极氧化处理的决定性因素[43]。 电解质溶液的组成强烈地影响阳极氧化成膜过程及膜层性能。 电解质溶液的组成不同, 阳极氧化现象如火花放电形成和移动的速度、 保持连续火花的电位以及形成固定火花的趋势不同, 所得膜层的颜色、 质地(如微孔尺寸和粗糙度)、 厚度、 化学组成以及电化学性质等也不同[44]。 Gulbrandsen[45]根据电解质对高纯镁合金阳极的影响, 而将电解质分成4类:
1) 强钝化剂, 如氟化物和铬酸盐;
2) 中等钝化剂, 如氢氧化物、 碳酸盐、 硼酸盐和磷酸盐;
3) 中等腐蚀活化剂, 如硫酸盐、 醋酸盐和硝酸盐;
4) 强点蚀催化剂, 如氯化物、 溴化物和可能的高氯化物。
从表3可以看出, 镁合金阳极氧化所用的电解质一般为镁的强钝化剂和中等钝化剂, 应用较多的是氢氧化物, 也有的配方中采用了中等腐蚀活化剂如硫酸盐[19, 37]。
Mg-H2O体系φ―pH图如图3[46]所示。 从图3可看出, 只有在碱性溶液中镁才能稳定存在。 张永君对镁阳极氧化成膜过程热力学可能性作进一步分析, 得出pH≥11.475是镁阳极氧化膜稳定存在的pH条件[47], 这是镁合金阳极氧化电解液中常加入碱金属氢氧化物的原因。

图3 Mg-H2O体系φ―pH图(Pourbaix图)
Fig.3 φ―pH diagram of Mg-H2O system (Pourbaix)
在酸性含氟的溶液中, 镁阳极氧化膜也能生长, 这是由于形成了难溶解的MgF2, 阻止了镁的腐蚀所致。 在选择电解质时, 基本原则之一是选用那些能与镁形成稳定沉淀的物质, 而氟化物、 氢氧化物和磷酸盐均能满足这一条件。 除了以上3类物质外, 还常用硅酸盐[11, 30, 31, 36, 39-41, 43, 48-54]和铝酸盐(或碱性中加入氢氧化铝)[14, 15, 27, 37, 55-58]。
各种电解质在镁阳极氧化中所起的作用, 目前研究得还不深入, 文献报道得也少。 碱金属氢氧化物除了能调节溶液的pH值外, 还具有提高氧化层硬度的作用[38]。 另外溶液中碱金属氢氧化物的用量直接与击穿电压有关, 用量太低时, 形成阳极氧化层的电压太高, 且氧化层太粗糙; 含量高时, 电解电压不会达到理想水平[31] 。 羧酸盐与氧化层的密度有关, 硼酸盐与膜层的厚度和耐磨性有关[36, 40]。 过氧化物能降低形成理想膜层的电压[32, 35]。 例如, 5%氨水+0.05mol/L NaNH4-HPO4+0.1mol/L Na2O2直流210V产生的膜层与不加Na2O2 300V时产生的膜层类似[35]。 阳极氧化时, 加入氨水或二亚乙基三胺等有机胺物质可以使外加电压达到很高而不产生火花, 具体能达到的电压与所用的氨水或二亚乙基三胺用量和其它电解质浓度有关[32, 35, 59]。 其它的胺类物质如六亚甲基四胺[20, 24]、 羟胺[8]和三乙胺[60]均有使用。
电解液中加入铝盐(Al(OH)3或AlO-2、 Al(NO3)3)后, 能使氧化膜中含有Al2O3[58, 61]或形成具有尖晶石结构的MgAl2O4[27, 55-57]。 另外, 电解液中加入Al(NO3)3可使氧化膜更均匀和致密, 但降低了氧化膜的厚度[61]。 Sato[62]则研究了在DOW17中添加Al2(SO4)3的作用, 发现添加Al2(SO4)3不仅使得阳极氧化处理后腐蚀速度降低, 而且大大抑制了电偶腐蚀的发生。 尖晶石缺位较少, 因而离子不易通过, 具有较好的抗腐蚀和耐磨性能[37]。
李建中等[63]研究了不同含磷电解液在微弧氧化过程中对成膜速度、 膜的结构形貌及膜的组成影响, 结果表明, 磷元素的大化学计量比可以提高微弧氧化成膜速度, 降低氧化膜孔隙率, 提高致密性, 增强与基体的结合力, 并改变了氧化膜的组成。 同时, 分子的聚合状态也影响其成膜速度和氧化膜的孔隙率, 链状聚合物可提高氧化膜成膜速度及氧化膜的致密性, 而环状聚合物对其影响较小。
3 镁合金阳极氧化成膜机理
镁合金阳极氧化过程与铝合金有很大不同, 不仅所用的电解液不同, 而且由于镁更活泼因而氧化膜生长机制更复杂[64]。 目前, 镁的阳极氧化不像铝那样成熟和容易, 工业化和商品化水平远不及铝阳极氧化。 两种金属阳极氧化工艺的具体区别如表4所列。
当镁合金氧化时, 由于火花放电导致样品表面的温度很高, 超过1000℃[11, 27], 在这么高的温度下, 金属元素的氧化可以看作高温氧化。 在高温反应过程中, 金属氧化物的热力学稳定性与反应过程进行的可能性和平衡有很大的关系, 各种同类型化合物的稳定性可用它们的标准生成吉布斯自由能来比较。 比较各种氧化物的稳定性时, 由于每一氧化物分子所含的氧原子数各不相同, 因此其生成吉布斯自由能不能以1mol氧化物为单位, 而应以1mol氧为单位。 另外, 纯镁的机械强度低, 不做结构材料使用。 在工业上, 纯镁除了少部分用于化学工业、 仪表制造及军事工业外, 主要用于制造镁合金及生产镁铝合金的合金元素。 镁中加入合金元素后, 使得氧化膜的生长机制更加复杂。
首先, 从热力学方面来看, 镁合金中的其它元素由于它们的氧化物的吉布斯自由生成能一般比MgO大(稀土元素除外), 因此镁首先氧化, 合金元素在合金/氧化膜下开始富集, 富集层的厚度一般为几十纳米[65]。 当合金元素富集到一定浓度后, 即开始氧化, 形成氧化物[51, 65-67]。 镁中加入合金元素Al、 Mn、 Zn、 W和Cu的理论富集量如图4所示。
表4 铝合金和镁合金阳极氧化工艺比较[23]
Table 4 Comparison of anodizing processes of aluminium alloys and magnesium alloys[23]
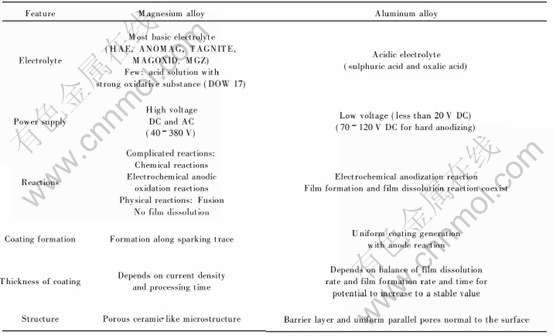
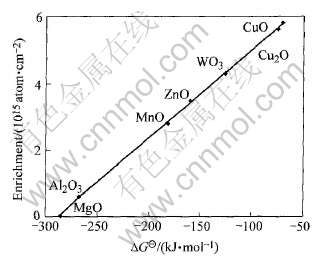
图4 含有1%~5%铝的镁合金预计的富集量[65]
Fig.4 Predicted enrichment of Mg alloys containing 1%-5% aluminium[65]
合金元素的富集现象首先是在研究铝合金的阳极氧化时发现的[68, 69]。 当合金元素形成氧化物的吉布斯自由生成能高于氧化铝的自由能时, 发生富集。 但是富集现象并未发生在Ta-Cu合金[70]、 Nb-Cu合金[71]和Zr-W[72]合金阳极氧化上。 因此合金阳极氧化时, 仅考虑热力学是不够的, 其它因素如膜的结构、 合金中当地原子排列、 合金/膜表面形成的膜物质量与消耗的合金量的关系, 可能与一些合金体系中发生合金富集有关[65]。
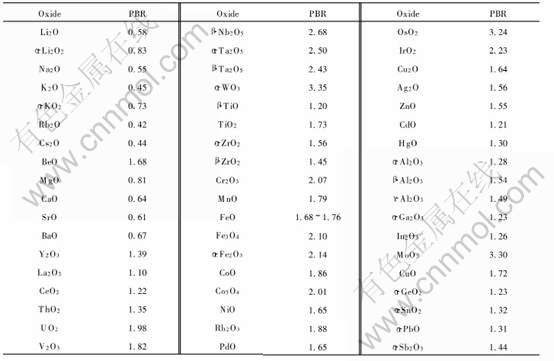
其次, 当镁中的合金元素开始氧化后, 由于它们氧化物的PBR值(Pilling-Bedworth Ratio)与MgO的不同, 使得氧化膜的结构发生变化。 常见合金元素氧化物的PBR值如表5所列。
由表5可见, Li2O等氧化物的PBR值比MgO小, 因此对于含有以上合金元素的镁合金来说, 当合金元素氧化后, 由于合金氧化物的PBR值比MgO小, 因此氧化膜中可能出现空隙或空位[73, 74]。 而对于含有诸如铝元素的镁合金来说, 由于Al2O3等氧化物的PBR值大于1, 因此当铝氧化后, 氧化膜内存在压应力使氧化膜的受力情况发生改变。
另外, 合金元素氧化成离子后, 由于它们和氧的结合能与Mg2+-O不同, 使得离子的移动速度与镁离子不同[75, 76], 导致氧化膜的成分发生变化, 如Mg2+的移动速度比Al3+快3倍[75], 而Li+的移动速度是Al3+的8倍[76]。
镁阳极氧化, 包含膜的生长和基体金属溶解两个过程。
镁作为阳极, 膜的生长以基体金属的消耗为前提条件。 假定“电流效率为百分之百”以及“阳极膜由完全均匀、 致密的MgO组成”, 在法拉第区, 消耗掉的基体金属平均膜厚为
![]()
式中 J为电流密度, mA/cm2; t为电解时间, s。
表5 氧化物的PBR值[78]
Table 5 PBR values of oxides[78]
所形成的膜层的平均膜厚为
![]()
由于膜层厚度以基体金属实际表面为基准, dc、 df的差异必然导致阳极试样的总体减薄[47]。 而实际情况是, 阳极氧化后, 试样一般会增厚。 薛文斌等[15]为研究ZM5铸造镁合金微弧氧化膜的生长规律, 使用精度为0.01mm的螺旋测微计测定氧化前后样品尺寸变化, 结果为氧化后的试样增厚了, 最大约为45μm。 理论与实际的差异, 一方面是阳极氧化时, 由于析出气体, 电流效率不可能百分之百, 同时膜层中存在孔隙、 缺陷; 另一方面, 阳极氧化时, 氧化膜内部电场强度较大, 可达到106V/cm[64, 77], 在这么高的电场作用下, O2-向氧化膜内部迁移, 溶液中的其它阴离子也会向阳极迁移并进入膜中[56, 64]。 Ono等[64]假定膜完全由MgO组成, 通过测量膜厚, 再根据法拉第公式计算, 电流效率约为400%, 这说明除了一般的电化学反应物质外, 电解液中的其它物质也进入了膜中。
如果阳极氧化膜完全由MgO组成, 其为n型半导体[78], 电子电流可以依靠相应的机构在导带内流动。 假如除了金属被氧化成正价离子的阳极反应外, 没有其它阳极反应能够发生, 于是阳极反应的进行必须经历这种传质过程: 离子通过钝化膜而生成新的钝化膜层。 可能是由于带正电荷的金属离子从钝化膜与金属界面穿过钝化膜向外移动, 在钝化膜外表面形成新的钝化膜; 也可能是带负电荷的O2-离子从钝化膜外表面向金属方向移动, 在钝化膜的内侧形成新的钝化膜层; 也可以是既有金属离子从钝化膜内侧向外移动, 也有O2-离子从膜的外侧向内移动, 而于钝化膜中某深度处形成新的钝化膜层[79]。
研究阳极氧化扩散规律的方法主要有以下几种: 1) Xe125作为示踪原子, 它的厚度通过β射线能谱仪测定[80, 81]; 2) Rn222作为示踪原子, 厚度通过测量发出的α离子能量损失来确定[82]; 3) 化学切片结合放射性测量来确定Rn222, Xe125, Kr85, Ar41的位置[83, 84]。 以上方法均需要放射性示踪原子或切片技术。 Brown等[85]使用了一种确定标识物位置的方法: Rutherford scattering。 这种方法是得到靠近膜层表面大约1μm地方元素厚度分布信息最有效的方法之一[69], 但这种方法也需要注入惰性原子。 对于阀金属的氧化扩散规律, 铝合金阳极氧化研究得较多。 电解液对金属离子和氧离子迁移系数影响很大, 在电流密度为20A/m2、 温度为296K及5%五硼酸铵水溶液中, Al3+的迁移系数为0.39; 而在电流密度为1~100A/m2这个很宽的范围内、 温度298 K及50g/L五硼酸铵混合溶液中(95%乙二醇、 5%水), 其迁移系数为0.6[85]。 以上方法比较准确, 但都必须注入惰性原子或使用放射性原子, 费用高、 程序复杂。 薛文斌等[15, 86, 87]使用一种简单的方法来研究氧化膜的生长机制, 即通过测量氧化膜的总厚度以及试样氧化前后的尺寸变化, 从而得出向外生长和向里生长的厚度。 这种方法虽然简单, 只需要涡流测厚仪和螺旋测微计, 但误差比较大。
研究离子扩散规律的示意图如图5所示。 图中虚线为氧化前注入的惰性原子如Xe原子的位置, 它由于在氧化过程中不会移动, 因此作为基准线。 图5(a)中的虚线到下面实线的距离为氧化前形成的很薄的氧化膜层。 图5(b)表示溶液中除O2-离子以外的阴离子如Y-离子在氧化膜增长过程中向里迁移的示意图, b∶a的值表示Y- 离子与O2-离子迁移速率的比值。 阴离子的移动速度一般比O2-慢, 如PO3-4的移动速度只有O2-的0.51[88], 但是F-的速度却比O2-快[89]。

图5 溶液中的阴离子和基体中的阳离子扩散示意图[77]
Fig.5 Schematic diagrams illustrating incorporation of foreign species derived from electrolyte anions and alloy substrates into anodic films
图5(c)所示为基体中除Mg2+离子以外的阳离子如X+离子在氧化膜增长过程中向外迁移的示意图, b∶a的值表示X+离子与Mg2+离子迁移速率的比值。 图5(d)所示为基体中除Mg2+离子以外的阳离子如X+离子在氧化膜增长过程中向外迁移的速度比Mg2+快, 从而在氧化膜表面形成一层富X元素的膜层(见图中的粗实线)。
关于击穿机制, 已提出了许多理论, 有氧化物裂缝形成机制[90]、 雪崩增殖[91, 92]机制。 Van等[28]认为氧化膜中出现了裂缝, 成为薄弱部位, 当外加电压大于薄弱部位的击穿电压时, 该部位首先被击穿。 裂缝机制能定性解释击穿电压与溶液电阻的关系, 但是它不能解释为什么击穿电压与膜形成过程、 以前存在的击穿和电极表面形貌无关, 因为这些因素影响裂缝和裂纹的种类和数量[93]。 Klein[94]于1972年提出了击穿由随机事件引发, 例如某一点雪崩的连续, Kadary等[95]用该理论成功地解释了击穿现象。
4 结语
经过半个多世纪的探索, 镁合金阳极氧化取得了很大进展, 但是有一些方面需要完善[96]:
1) 由于镁的活性高, 因此大多数阳极氧化电解液中使用了对环境和人身有害的物质, 如铬酐、 氟化物或磷酸盐等。 在环境保护越来越重要的今天, 为了顺应时代发展的要求, 开发对环境没有污染的电解液配方, 非常有必要。
2) 电参数、 溶液温度等对镁合金氧化膜的耐蚀性影响很大。 随着科技进步, 微弧氧化电源发展的主要成果是频率不断提高, 并且包含正脉冲电源和负脉冲电源, 正、 负脉冲的占空比在一定范围内可分别单独调节。 目前研究电参数尤其是频率、 占空比、 电流密度以及终电压对氧化膜性能的影响还不多。
3) 阀金属阳极氧化膜的形成, 包括被氧化后的金属离子向外扩散和氧离子向里扩散两个过程。 相对于铝合金阳极氧化而言, 镁合金阳极氧化由于研究得较晚, 氧化膜的生长机制研究得还不彻底, 这方面仍须深入。
4) 阳极氧化是提高镁合金耐蚀性的一种有效方法, 但是它需要耗电, 这是限制该方法普遍应用的一个因素, 尤其是在能源日益匮乏的今天, 因此测量阳极氧化电流效率非常有必要, 它能为工业生产提供指导意义。
REFERENCES
[1]Funatani K. Emerging technology in surface modification of light metals[J]. Surface and Coatings Technology, 2000, 133-134: 264-272.
[2]Froes F H, Eliezer D, Aghion E. The science, technology, and applications of magnesium[J]. JOM, 1998, 5(9): 30-34.
[3]Decker R F. The renaissance in magnesium[J]. Advanced Materials and Processes, 1998, 9: 31-33.
[4]师昌绪, 李恒德, 王淀佐, 等. 加速我国金属镁工业发展的建议[J]. 材料导报, 2001, 15(4): 5-7.
SHI Chang-xu, LI Heng-de, WANG Dian-zuo, et al. A proposal on accelerating development of metallic magnesium industry in China[J]. Materials Review, 2001, 15(4): 5-6.
[5]曾荣昌, 柯伟, 徐永波, 等. Mg合金的最新发展及应用前景[J]. 金属学报, 2001, 37(7): 673-685.
ZENG Rong-chang, KE Wei, XU Yong-bo, et al. Recent development and application of magnesium alloys[J]. Acta Metalllurgica Sinica, 2001, 37(7): 673-685.
[6]余刚, 刘跃龙, 李瑛, 等. Mg合金的腐蚀与防护[J]. 中国有色金属学报, 2002, 12(6): 1087-1098.
YU Gang, LIU Yue-long, LI Ying, et al. Corrosion and protection of magnesium alloys[J]. The Chinese Journal of Nonferrous Metals, 2002, 12(6): 1087-1098.
[7]Gray J E, Luan B. Protective coatings on magnesium and its alloys―A critical review[J]. Journal of Alloys and Compounds, 2002, 336: 88-113.
[8]Ostrovsky, Ilya. Method of anodizing of magnesium and magnesium alloys and producing conductive layers on an anodized surface [P]. US Patent: 20030000847, 2003-01-02.
[9]Survila E. Modern anodizing[J]. Trans IMF, 1984, 62: 45-47.
[10]McNeill W. The Cr-22 coating for magnesium[J]. Metal Finishing, 1955, 53(12): 57-59.
[11]Zozulin A J, Bartak D E. Anodized coatings for magnesium alloys[J]. Metal Finishing, 1994, 92(3): 39-44.
[12]Dittrich K H, Krysmann W, Kuaze P, et al. Structure and properties of ANOF layers[J]. Crystal Res and Technol, 1984, 19(1): 93-99.
[13]Xue W, Deng Z, Lai Y, et al. Analysis of phase distribution for ceramic coatings formed by microarc oxidation on aluminum alloy[J]. J Am Ceram Soc, 1998, 81(5):1365-68.
[14]薛文斌, 来永春, 邓志威, 等. 镁合金微等离子体氧化膜的特性[J]. 材料科学与工艺, 1997, 5(2): 89-92.
XUE Wen-bin, LAI Yong-chun, DENG Zhi-wei, et al. The properties of coating formed by microplasma oxidation on magnesium alloy[J]. Material Science & Technology, 1997, 5(2): 89-92.
[15]薛文斌, 邓志威, 来永春, 等. ZM5镁合金微弧氧化膜的生长规律[J]. 金属热处理学报, 1998, 19(3): 42-45.
XUE Wen-bin, DENG Zhi-wei, LAI Yong-chuan, et al. Growth regularity of ceramic film in microarc oxidation on cast magnesium alloy[J]. Transactions of Metal Heat Treatment, 1998, 19(3): 42-45.
[16]Yerokhin A L, Nie X, Leyland A, et al. Plasma electrolysis for surface engineering[J]. Surface and Coatings Technology, 1999, 122: 73-79.
[17]Kuhn A. Plasma anodizing of magnesium alloys[J]. Metal Finishing, 2003, 101(9): 44-50.
[18]Beauvir, Jacques. Oxidizing electrolytic method for obtaining a ceramic coating at the surface of a metal [P]. US Patent: 6808613, 2004-10-26.
[19]Schmeling, Edith L, Roschenbleck, et al. Method of preparing the surfaces of magnesium and magnesium alloys[P]. US Patent: 4976830, 1990-12-11.
[20]Schmeling, Edith L, Roschenbleck, et al. Method of producing protective coatings that are resistant to corrosion and wear on magnesium and magnesium alloys[P]. US Patent: 4978432, 1990-12-18.
[21]Xiang Y, Hu W, Liu X, et al. A study on surface state during the pretreatment of electroless nickel plating on magnesium alloys[J]. Trans IMF, 2001, 79(1): 27-29.
[22]霍宏伟, 李瑛, 王福会. AZ91D镁合金化学镀镍[J]. 中国腐蚀与防护学报, 2002, 22(1): 14-16.
HUO Hong-wei, LI Ying, WANG Fu-hui. Electroless nickel plating of AZ91D magnesium alloys[J]. Journal of Chinese Society for Corrosion and Protection, 2002, 22(1): 14-16.
[23]SHI Z. The corrosion performance of anodized magnesium alloys[D]. Queensland: The University of Queensland, Brisbane, Australia, 2004. 1-23.
[24]Kurze, Peter, Banerjee, et al. Method of producing oxide ceramic layers on barrier layer-forming metals and articles produced by the method[P]. US Patent: 5385662, 1995-01-31.
[25]Burger F J, Wu J C. Dielectric breakdown in electrolytic capacitors[J]. J Electrochem Soc, 1971, 118(12): 2039-2042.
[26]李宝东, 申泽骥. 镁合金铸件表面处理技术现状[J]. 材料保护, 2002, 35(4): 1-3.
LI Bao-dong, SHEN Ze-ji. Surface treatment technique of magnesium alloy castings[J]. Materials Protection, 2002, 35(4): 1-3.
[27]Khaselev O, Weiss D, Yahalom J. Structure and composition of anodic films formed on binary Mg-Al alloys in KOH-aluminate solutions under continuous sparking[J]. Corrosion Science, 2001, 43: 1295-1307.
[28]Van T B, Brown S D, Wirtz G P. Mechanism of anodic spark deposition[J]. Am Ceram Soc Bulletin, 1977, 56(6): 563-566.
[29]刘风岭, 骆更新, 毛立信. 微弧氧化与材料表面陶瓷化[J]. 材料保护, 1998, 31(3): 22-24.
LIU Fen-ling, LUO Geng-xin, MAO Li-xin. Micro-arc oxidation and ceramic-like coating on material surface[J]. Materials Protection, 1998, 31(3): 22-24.
[30]蒋百灵, 张淑芬, 吴国建. 镁合金微弧氧化陶瓷层耐蚀性的研究[J]. 中国腐蚀与防护学报, 2002, 22(5): 300-303.
JIANG Bai-ling, ZHANG Shu-fen, WU Guo-jian. The study of the corrosion resistance of the ceramic coatings formed by micro-arc oxidation on the Mg-base alloy[J]. Journal of Chinese Society for Corrosion and Protection, 2002, 22(5): 300-303.
[31]Bartak, Duane E, Lemieux, et al. Hard anodic coating for magnesium alloys[P]. US Patent: 5470664, 1995-11-28.
[32]Barton, Francis T, Macculloch, et al. Anodization of magnesium and magnesium-based alloys[P]. US Patent: 6280598, 2001-08-28.
[33]Evangelides H A. Method of electrolytically coating magnesium and electrolyte therefor[P]. US Patent: 2723952, 1955-11-15.
[34]Dow Chemical Co. Bath for and method of producing a corrosion resistant coating upon light metals[P]. GB Patent: 762195, 1956-11-28.
[35]Barton, Francis T. Anodization of magnesium and magnesium based alloys[P]. US Patent: 5792335, 1998-08-11.
[36]Kobayashi, Waichi, Uehori, et al. Anodizing solution for anodic oxidation of magnesium or its alloys[P]. US Patent: 4744872, 1988-05-17.
[37]Kobayashi, Waichi, Takahata, et al. Aqueous anodizing solution and process for coloring article of magnesium or magnesium-base alloy[P]. US Patent: 4551211, 1985-11-05.
[38]Habermann, Clarence E, Garrett, et al. Anticorrosive coated rectifier metals and their alloys[P]. US Patent: 4668347, 1987-05-26.
[39]Kozak, Otto. Anti-corrosive coating on magnesium and its alloys[P]. US Patent: 4184926, 1980-01-22.
[40]Kozak, Otto. Method of coating articles of magnesium and an electrolytic bath therefore[P]. US Patent: 4620904, 1986-11-04.
[41]Bartak, Duane E, Lemieux, et al. Two-step electrochemical process for coating magnesium alloys[P]. US Patent: 5264113, 1993-11-23.
[42]Dolan, Shawn E. Light metal anodization[P]. US Patent: 20030070936, 2003-04-17.
[43]张永君, 严川伟, 王福会, 等. 镁及镁合金环保型阳极氧化电解液及其工艺[J]. 材料保护, 2002, 35(3): 39-41.
ZHANG Yong-jun, YAN Chuan-wei, WANG Fu-hui, et al. Environmental-friendly bath solution and process for anodization of magnesium and its alloys[J]. Materials Protection, 2002, 35(3): 39-41.
[44]张永君, 严川伟, 楼翰一, 等. Mg及其合金的阳极氧化技术进展[J]. 腐蚀科学与防护技术, 2001, 13(4): 214-217.
ZHANG Yong-jun, YAN Chuan-wei, LOU Han-yi, et al. Progress on anodizing technology for magnesium and its alloys[J]. Corrosion Science and Protection Technology, 2001, 13(4): 214-217.
[45]Gulbrandsen E. Anodic behaviour of Mg in HCO-3/CO2-3 buffer solutions. Quasi-steady measurements[J]. Electrochimica Acta, 1992, 37(8): 1403-1412.
[46]杨熙珍, 杨武. 金属腐蚀电化学热力学电位―pH图及其应用[M]. 北京: 化学工业出版社, 1991.
YANG Xi-zhen, YANG Wu. Potential―pH diagram of electrochemistry and thermodynamics on metal corrosion and its applications[M]. Beijing: Chemical Industry Press, 1991.
[47]张永君. 镁及镁合金环保型阳极氧化表面改性技术研究[D]. 沈阳: 中国科学院金属研究所, 2003. 66-88.
ZHANG Yong-jun. Study on environmentally friendly anodizing surface modification for magnesium and its alloys[D]. Shenyang: Institute of Metal Research, Chinese Academy of Sciences, 2003. 66-88.
[48]蒋百灵, 张淑芬, 吴国建, 等. 镁合金微弧氧化陶瓷层显微缺陷与相组成及其耐蚀性[J]. 中国有色金属学报, 2002, 12(3): 454-457.
JIANG Bai-ling, ZHANG Shu-fen, WU Guo-jian, et al. Microflaw and phase constitution of ceramic coating formed by micro-arc oxidation on magnesium alloys and their influence on corrosion-resistance[J]. The Chinese Journal of Nonferrous Metals, 2002, 12(3): 454-457.
[49]郝建民, 陈宏, 张荣军, 等. 微弧氧化和阳极氧化处理镁合金的耐蚀性对比[J]. 材料保护, 2003, 36(1): 20-21.
HAO Jian-min, CHEN Hong, ZHANG Rong-jun, et al. Corrosion resistances of magnesium alloys by micro-arc oxidation and anodic oxidation[J]. Materials Protection, 2003, 36(1): 20-21.
[50]Zhang Y, Yan C, Wang F, et al. Study on the environmentally friendly anodizing of AZ91D magnesium[J]. Surface and Coatings Technology, 2002, 161: 36-43.
[51]Mato S, Alcala G, Skeldon P, et al. High resistivity magnesium-rich layers and current instability in anodizing a Mg/Ta alloy[J]. Corrosion Science, 2003, 45: 1779-1792.
[52]Fukuda H, Matsumoto Y. Effects of Na2SiO3 on anodization of Mg-Al-Zn alloy in 3 M KOH solution[J]. Corrosion Science, 2004, 46: 2135-2142.
[53]Birss V, Xia S, Yue R, et al. Characterization of oxide films formed on Mg-based WE43 alloy using AC/DC anodization in silicate solutions[J]. J Electrochem Soc, 2004, 151(1): B1-B10.
[54]Xia S J, Yue R, Rateick, et al. Electrochemical studies of AC/DC anodized Mg alloy in NaCl solution[J]. J Electrochem Soc, 2004, 151(3): B179-B187.
[55]Khaselev O, Yahalom J. Constant voltage anodizing of Mg-Al alloys in KOH-Al(OH)3 solution[J]. J Electrochen Soc, 1998, 145(1): 190-193.
[56]Khaselev O, Weiss D, Yahalom J. Anodizing of pure magnesium in KOH-aluminate solutions under sparking[J]. J Electrochem Soc, 1999, 146(5): 1757-1761.
[57]Khaselev O, Yahalom J. The anodic behavior of binary Mg-Al alloys in KOH-aluminate solutions[J]. Corrosion Science, 1998, 40(7): 1149-1160.
[58]Verdier S, Boinet M, Maximovitch S, et al. Formation, structure and compositions of anodic films on AM60 magnesium alloy obtained by DC plasma anodizing[J]. Corrosion Science, 2005, 47: 1429-1444.
[59]罗胜连, 张涛, 周海晖, 等. 有机胺对镁合金阳极氧化的影响[J]. 中国有色金属学报, 2004, 14(4): 691-696.
LUO Sheng-lian, ZHANG Tao, ZHOU Hai-hui, et al. Effects of organic amine on anodizing of magnesium alloys[J]. The Chinese Journal of Nonferrous Metals, 2004, 14(4): 691-696.
[60]Asoh H, Ono S. Anodizing of magnesium in amine-ethylene electrolyte[J]. Materials Science Forum, 2003, 419-422: 957-962.
[61]Hsiao H Y, Tsai W T. Characterization of anodic films formed on AZ91D magnesium alloy[J]. Surface and Coatings Technology, 2005, 190: 299-308.
[62]Sato F, Asakawa Y. Corrosion behavior of magnesium alloy AZ91D anodized in aluminum sulfate added solution[J]. Journal of Japan of Light Metals. 1994, 44(2): 104-109.
[63]李建中, 邵忠财, 田彦文, 等. 不同含磷电解液在微弧氧化过程中的作用[J]. 中国腐蚀与防护学报, 2004, 24(4): 222-225.
LI Jian-zhong, SHAO Zhong-cai, TIAN Yan-wen, et al. The action of the forms of P element on the process of the microarc oxidation[J]. Journal of Chinese Society for Corrosion Protection, 2004, 24(4): 222-225.
[64]Ono S, Asami K, Osaka T, et al. Structure of anodic films formed on magnesium[J]. J Electrochem Soc, 1996, 143(3): L62-L63.
[65]Bonilla F A, Berkani A, Skeldon P, et al. Enrichment of alloying elements in anodized magnesium alloys[J]. Corrosion Science, 2002, 44: 1941-1948.
[66]Bonilla F A, Berkani A, Liu Y, et al. Formation of anodic films on magnesium alloys in an alkaline phosphate electrolyte[J]. J Electrochem Soc, 2002, 149(1): B4-B13.
[67]Abulsain M, Berkani A, Bonilla F A, et al. Anodic oxidation of Mg-Cu and Mg-Zn alloys[J]. Electrochimica Acta, 2004, 49: 899-904.
[68]Strehblow H H, Doherty C J. Examination of aluminum copper films during anodic oxidationⅠ.Corrosion studies[J]. J Electrochem Soc, 1978, 125(1): 30-33.
[69]Strehblow H H, Melliar-Smith C M, Augustyniak W M. Examination of aluminum-Copper films during the galvanostatic formation of anodic oxide Ⅱ: Rutherford backscattering and depth profiling[J]. J Electrochem Soc, 1978, 125(6): 915-919.
[70]Mato S, Thompson G E, Skeldon P, et al. Enrichment of alloying elements beneath anodic oxides: investigation of Ta-1.5at.% Cu alloy[J]. Corrosion Science, 2001, 43: 993-1002.
[71]Mato S, Skeldon P, Thompson G E, et al. Behaviour of copper and generation of oxygen during anodizing of Nb-Cu alloys[J]. Surface and Interface Analysis, 2000, 29: 895-902.
[72]Habazaki H, Shimizu K, Skeldon P, et al. Incorporation of tungsten species into the anodic film on Zr-2.7 atom percent W alloy[J]. J Electrochem Soc, 1997, 144(10): 3492-3495.
[73]Zhou X, Thompson G E, Skeldon P, et al. Film formation and detachment during anodizing of Al-Mg alloys[J]. Corrosion Science, 1999, 41:1599-1613.
[74]Thompson G E, Skeldon P, Wood G C, et al. Elastic recoil detection analysis (EDRA), RBS and TEM study of barrier film formation on Al-4.5at.%Mg-0.005at.%Cu alloy[J]. Surface and Interface Analysis, 1999, 27: 57-62.
[75]Liu Y, Skeldon P, Thompson G E, et al. Anodic film growth on an Al-21at.%Mg alloy[J]. Corrosion Science, 2002, 44: 1133-1142.
[76]Tzoganakou K, Skeldon P, Thompson G E, et al. Mobility of lithium ions in anodic alumina formed on an Al-Li alloy[J]. Corrosion Science, 2000, 42: 1083-1091.
[77]Habazaki H, Shimizu K, Skeldon P, et al. Effects of alloying elements in anodizing of aluminium[J]. Trans IMF, 1997, 75(1): 18-23.
[78]李美栓. 金属的高温腐蚀[M]. 北京: 冶金工业出版社, 2001.
LI Mei-shuan. Metal Corrosion at High Temperatures[M]. Beijing: Metallurgical Industry Press, 2001.
[79]曹楚南. 腐蚀电化学[M]. 北京: 化学工业出版社, 1995.
CAO Chu-nan. Corrosive Electrochemistry[M]. Beijing: Chemical Industry Press, 1995.
[80]Davies J A, Pringle J P S, Graham R L, et al. A radiotracer studies of anodic oxidation[J]. J Electrochem Soc, 1962, 109(10): 999-1001.
[81]Davies J A, Domeij B, Pringle J P S, et al. The migration of metal and oxygen during anodic film formation[J]. J Electrochem Soc, 1965, 112(7): 675-680.
[82]Davies J A, Domeij B. The use of α-spectroscopy for studying anodic oxidation[J]. J Electrochem Soc, 1963, 110(7): 849-852.
[83]Pringle J P S. A very precise sectioning method for measuring concentration profiles in anodic tantalum oxide[J]. J Electrochem Soc, 1972, 119(4): 482-491.
[84]徐源, Thompson G E, Wood G C. 铝阳极氧化膜形成过程中离子迁移的规律及对膜形态的影响[J]. 中国腐蚀与防护学报, 1988, 8(1): 1-13.
XU Yuan, Thompson G E, Wood G C. Effects of ionic transport regularity on coating configuration during anodic coating formation on aluminum alloy[J]. Journal of Chinese Society for Corrosion Protection, 1988, 8(1): 1-13.
[85]Brown F, Machintosh W D. The use of Rutherford Backscattering to study the behaviour of ion-implanted atoms during anodic oxidation of aluminum: Ar, Kr, Xe, Rb, Cs, Cl, Br, and I[J]. J Electrochem Soc, 1973, 120(8): 1096-1102.
[86]薛文斌, 邓志威, 来永春, 等. 铝合金微弧氧化膜尺寸变化规律[J]. 中国有色金属学报, 1997, 7(3): 140-143.
XUE Wen-bin, DENG Zhi-wei, LAI Yong-chun, et al. Size change of LY12 aluminum alloy in process of microarc oxidation[J]. The Chinese Journal of Nonferrous Metals, 1997, 7(3): 140-143.
[87]薛文斌, 邓志威, 张通和, 等. 铸造镁合金微弧氧化机理[J]. 稀有金属材料与工程, 1999, 28(6): 353-356.
XUE Wen-bin, DENG Zhi-wei, ZHANG Tong-he, et al. Microarc oxidation mechanism of a cast magnesium alloy[J]. Rare Metal Materials and Engineering, 1999, 28(6): 353-356.
[88]Shimizu K, Brown G M, Habazaki H, et al. Incorporation and migration of Cr3+ and PO3-4 species in anodic alumina[J]. Corrosion Science, 1999, 41: 1971-1976.
[89]Felhosi I, Habazaki H, Shimizu K, et al. Void formation and alloy enrichment during anodizing of aluminium alloys containing cadmium, indium and tin[J]. Corrosion Science, 1998, 40(12): 2125-2139.
[90]Axelrod N N, Schwartz N. Asymmetric conduction in thin film tantalum/tantalum oxide/metal structures: Interstitial and substitutional impurity effects and direct detection of flaw breakdown[J]. J Electrochem Soc, 1969, 116(4): 460-465.
[91]Vijh A K. Sparking voltages and side reactions during anodization of valve metals in terms of electron tunneling[J]. Corrosion Science, 1971, 11: 411-417.
[92]Fritzsche C R. Anodic growth, dielectric breakdown and carrier transport in amorphous SiO2 films[J]. J Phys Chem Solids, 1969, 30: 1885-1895.
[93]Ikonopisov S. Theory of electrical breakdown during formation of barrier anodic films[J]. Electrochimica Acta, 1977, 22: 1077-1082.
[94]Klein N. A theory of localized electronic breakdown in insulating films[J]. Adv Phys, 1972, 21: 605-644.
[95]Kadary V, Klein N. Electrical breakdownⅠ: During the anodic growth of tantalum pentoxide[J]. J Electrochem Soc, 1980, 127(1): 139-151.
[96]张荣发. 镁合金阳极氧化与电偶腐蚀的研究[D]. 沈阳: 中国科学院金属研究所, 2005. 1-30.
ZHANG Rong-fa. Study of anodization on galvanic corrosion in magnesium alloys[D]. Shenyang: Institute of Metal Research, Chinese Academy of Sciences, 2005. 1-30.
(编辑李艳红)
收稿日期: 2005-12-13; 修订日期: 2006-04-02
通讯作者: 张荣发, 教授; 电话: 0791-3801423; E-mail: rfzhang-10@hotmail.com

