���±��: 1004-0609(2005)07-1092-08
���Ͻ�LaNi5-xAlx���ӽṹ��Ӱ��
����1, ���»�1, �� ��2
(1. �Ĵ���ѧ ԭ������������о���, �ɶ� 610065; 2. �й����������о�Ժ, ���� 621900)
ժ Ҫ: ��ȫ����ˮƽ��, ���ڹ����ݶȽ����ܶȷ�����ȫ����������ƽ�沨����, �����˲�ͬAl����LaNi5-xAlx(x=0, 0.5, 1.0)�ľ���ṹ�� �ܴ��ṹ�� ״̬�ܶȵ������� ��LaNi5�ļ������: GGA Perdew96�ļ���������, �����������Ż��ṹ��������Ӱ�첻����; ��LaNi4Al���о�����Alԭ�������ȡ��3gλ��Niԭ��; ��LaNi4.5Al0.5���Ż��ṹ��ʵ����һ�¡� ����Al����������, ���������� ��������������, ���LaNi5, LaN4.5Al0.5��LaNi4Al, ������������-10.591�� -10.134eV���ߵ�-9.441eV, ��Ef�ϵ�̬�ܶȺͲ��϶�Ӧ�ĵ��±��������½�, ̬�ܶȴ�11.81�� 8.86���罵�͵�1.61eV/f.u.�� �����������LaNi4.5Al0.5���ܴ��ṹ�� ״̬�ܶ�ͼ��
�ؼ���: LaNi4Al; LaNi4.5Al0.5 ; ȫ����������ƽ�沨��(FLAPW) ��ͼ�����: TG139.7
���ױ�ʶ��: A
Alloying effects on electronic structures of LaNi5-xAlx
GAO Tao1, QI Xin-hua1, CHEN Bo2
(1. Institute of Atomic and Molecular Physics, Sichuan University, Chengdu 610065, China; 2. Chinese Academy of Engineering Physics, Mianyang 621900, China)
Abstract: Based on the generalized gradient approximation (GGA) of density of function and the full potential linearized augmented wave (FLAPW), the equilibrium structure , energy bands and density of states were calculated for LaNi5-xAlx. The results show that, For LaNi5, the calculation by GGA perdew96 is better than those of other methods, and the spin-polarization have no obvious effect on the optimized structure and energy. For LaNi4Al, the result indicates that nickel is most likely substituted by aluminum on the 3g site, and the optimized structure for LaNi4.5Al0.5 exactly agrees with the experimental results. With the increasing content of aluminum, the Fermi energy and density of states sharply decrease in the order from LaNi5 to LaNi4.5Al0.5and LaNi4Al, the Fermi Energy increases from -10.591, -10.134eV to -9.441eV, and the density of states (DOS) of Ef decreases from 11.81, 8.86 to 1.61eV/f.u.. The energy band structure and density of states for LaNi4.5Al0.5 were firstly calculated.
Key words: LaNi4Al; LaNi4.5Al0.5; electric structure; full-potential linearized augmented plane wave
LaNi5��AB5�ͻ������н�Ϊ����Ķ�Ԫ�������, ��ʵ��������϶��õ��˽�Ϊȫ����о��� ����, ���ֶ�Ԫ�Ͻ�ļ۸��, �ܶȴ�, ������ʱ��ƽ��ѹ����, �Լ��Էۻ���ѭ��������, ʹ��Ӧ���ܵ���һ�����ơ�
ͨ���Ͻ���[1, 2]������һ���̶��Ͽ˷�LaNi5������ϵ�����ȱ��, �ɴ˶���õĶ�Ԫ��������Ѿ�ʵ�û��� ��ΪAl���۴Ӽ۸� ��ѧ�ȶ��Ի��������ȽǶȽ�, �������ɴ��������ȶ�������Խ��, ������Alȡ������Ni�Ǵ����ѧ��[3-6]��ע�ķ���֮һ�� ������û���ܹ������ṹ���Ż������ �ԸúϽ�����Alԭ�ӵľ���ռλ�Լ���ͬ�������Խṹ������Ӱ��, Ŀǰ��û��һ�µĽ���[6-9]��
���ںϽ��е���״̬����ͬ����ϳɷֺ����ܵ��Ż����, ����ʵ��ɱ�������Ҫ�����塣 һ����ΪAlȡ���ĸ�λ������0~1.5֮��, ȡ����ľ���ṹ���Ա��������P6/mmm���䡣 ���ڳߴ�ЧӦ, Al����ȡ�������е�2c��3g��λ, ���ڵ�ʵ���о���Ϊ����ȡ����һ���������, ���Ǻ����ľ�ϸ�������������Ϊ���п���ȡ��3g��λ�� ���������[8]ͨ��cerius2���������ۼ���, ���ִ������ĽǶ�������������ȡ��, ��Al�����������ȡ�������ָ�λ�� ����, Ŀǰ���˶Ծ��и߶ȶԳ��Ե�LaNi5(P6/mmm)�ľ���ṹ�й������Ż��о�����, ��û������LaNi4.5Al0.5��LaNi4Al�ļ��νṹ���ܴ��ṹ�����������о�, ��Szajek��[6]�ڶ�LaNi4Al�Ľṹ�о���ֻ����ʵ�龧�������� ��Ȼ, ���ִ����Ľ������������ �������߽����ڹ����ݶȽ���(GGA: generalized gradient approximation)�ܶȷ�����ȫ����������ƽ�沨(FLAPW: full potential linearized augmented plane wave)����, ��ȫ����ˮƽ��, �����о���ͬ��������LaNi5�� LaNi4.5Al0.5�� LaNi4Al����ļ��νṹ�͵����ܶȡ� ״̬�ܶ�(Density of states)��, �����ۼ������, ����Al�����Ծ���ṹ�ߴ硢 �ܴ���״̬�ܶȵȵ�Ӱ��, �����ܼ��������õ�ģ���Լ����㷽��, �����˽�����������ϸ���ۡ�
1 ����ģ�������۷���
1.1 ����ṹ
LaNi5��һ�־���CaCu5�;���ṹ��ϡ������Ͻ�, ����Laռ1aλ, Niռ2c��3gλ, ��������ϵ, �ռ�ȺΪP6/mmm, ��ͼ1��ʾ�� ��ԭ����������:

����LaNi4Al, ����һ��LaNi5��������һ��Niԭ�Ӹ�λ��Alԭ��ȡ�����¡� Ϊ�˱Ƚ�����ȡ�������������, �������������Alԭ�ӷֱ�ȡ��LaNi5������2cλ��3gλ��һ��Niԭ�ӽṹ, ��������ģ��: ��LaNi4Al(3g)��LaNi4Al(2c), ��������������ڱ�1, ����La��ռ1aλ��
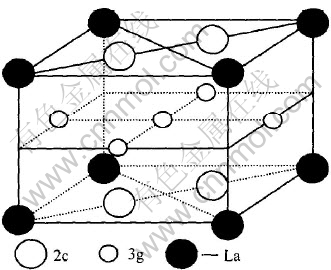
ͼ1 LaNi5�ľ����ṹ
Fig.1 Crystal structure of LaNi5
��1 ����LaNi4Alģ�͵����������
Table 1 Internal parameters of two models for LaNi4Al
����LaNi4.5Al0.5, ����Al�����Ƿ���, ����ͨ��������z�����һ������������2������, ������һ��Alԭ��ȡ���������е�һ��3g��λ��Niԭ�Ӻ�, ����Ի����Ӧ��Al��������, �����������һ����ʽ˵��:
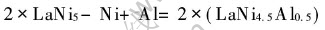
1.2���۷���
�����в���ȫ����ˮƽ�µ�ȫ����������ƽ�沨������ �÷����Լ��㾧��ĵ��ӽṹ���ȷ�ķ���֮һ, ͨ������������ϵ���г��������ƽ�沨�����������������ϵ��Kohn-Sham���̵Ļ�̬�����ܶȡ� �������ͱ���ֵ�� ���Ǵ����ܶȷ�������Ϊ�����ĵ�һ��ԭ������, ���Ƚ��������ӷ��̻�Ϊ�����ӷ���, ��Kohn-Sham����:
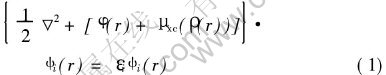
Ȼ��ͨ������������ƽ�沨�������Ե�����Schr?dinger(Kohn-Sham)���̽��м���, ͬʱҲ�����˹����ݶȽ������۽������ӵĽ����ܱ�ʾΪ�����ܶȼ����ݶȵĺ���, ��:
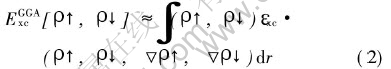
��ͬ���Ʒ������ڸ����в�ͬ�ļ���, ��������ʹ����Perdew96[7]��
������ƽ�沨(LAPW)�����Ծ����ڲ�ͬ����ѡ�ò�ͬ�Ļ������� ���IJ�����ͼ2��ʾ�ġ�Muffin-tin��ģ�͡� ����, ����Ϊ���ص���ԭ������, ����Ϊԭ�Ӽ�ļ�϶���� ����������, ��ԭ�ӵľ�������г���ֵij˻�:
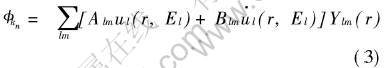
����������ȡƽ�沨չ��
��
������Kohn-Sham���̵Ľ�, ����ʾ������
ͼ2 Muffin-tinģ�͵Ļ���
Fig.2 Schematica of muffin-tin model [TS)] ���ֺ���������չ��:

�ڶ�Kohn-Sham������Ǣ�����ʱ, ����ģ�͵Ľ�����Ȼ���� ��Muffin-tin��ģ��, ��:

���ļ�����û����ܶȷ������۵ľ���ṹ��������WIEN2k, �������㷽���Ѱ����ڸó��������С�
2 ���������
�ڼ���Kohn-Sham����ʱ, ������Muffin-tinģ�ͶԲ�ͬ����ĵ���ʹ�ò�ͬ�IJ�����, ����La�� Ni��Alԭ�ӵ�RMT��Ϊ0.1058nm, RMT��Kmaxȡ7.0, ������г������չ��ָ��lȡ12.0; �ڽ���-����ܵļ�����, ����Perdew96����, ��ȡ-81.6eV���ڲ���Ӻͼ۵��ӷֿ�, �����ӵĵ�����̬�ֱ�Ϊ: La-5s25p65d16s2; Ni-3p63d84s2; Al-3s23p1�� �ڳ�ʼ��ʱ, ����Ԩ���еIJ���K����ȡΪ500, ���������巽���Զ����ռ���л���; �������������Ǣ�������������������ȿ���Ϊ1.36��10-3eV; ͬʱӦ��ţ����ѧ�����������Ӿ���LaNi4.5Al0.5�� LaNi4Alԭ��ƽ��λ�ý������Ż��� �����ЧӦͨ����������������, �����ж����ڲ���ӵļ��������ȫ����۵��ܶȷ���������
2.1 LaNi5�Ľṹ�Ż�����
LaNi5���и߶ȵĶԳ���, �����Ѿ��д�����ʵ����������ݿɲ�, ���Dz���ȫ����������ƽ�沨�����о��ı���������� ���������״�����WIEN2k������LaNi5�Ľṹ�����Ż��о�, �����������������������Ż��ṹ��Ӱ������������ �Ż��Ľṹ�������ڱ�2��
��2 LaNi5�Ż��ľ���ṹ����
Table 2 Optimized crystal structure of LaNi5
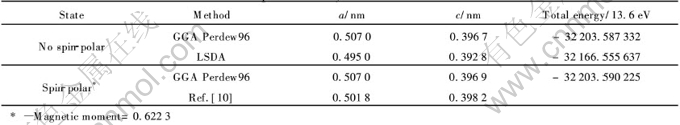
�ڲ�����������ЧӦ�������, ��2�м���������ʵ���������, GGA Perdew96������a�����Ϊ1.238%, c�����Ϊ0.376%, ��ʵ����ֵ�dz��ӽ�; LSDA������Դ�, ��������ֵƫ�ߺܶࡣ �ɼ�, GGA Perdew96���㷽��������������ֵ���, ����ں����ļ����н��������ַ�����
������������ЧӦ��, �ɱ�2��֪���������a�᷽��û�б仯, ��c�᷽����С�ı仯, ����0.0002nm; �ܵ�������û�п�����������ʱ����Լ4.08��10-2eV�� ���, ������������, ������������LaNi5����ṹ�ļ���Ӱ�첻���ԡ�
����LaNi5�Ĵ���, ʵ����Ϊ��˳���Ե�, ��û���ܹ��������ŵ�ʵ��ֵ�� ��Ŀǰ������Ч�ƺ�ƽ�沨�����ļ���������������LaNi5�����������Ե�, ��Hector��[11]�Ĵžؼ�����Ϊ1.1, ��Malik��[12]�ļ���ֵΪ0.69�� ���ĵļ������LaNi5Ϊ�����Ե�, ����Ĵž�Ϊ0.6223, ��Malik�ȵļ����������
2.2 LaNi4Al�Ľṹ�Ż�����
��LaNi4Al�ṹ�Ż�������, ��ԭ�ӵ�ƽ��λ��Ҳ�������Ż��� �Ż��õ��Ľṹ�������������ڱ�3��
��3 LaNi4Al�Ż���ľ���ṹ����
Table 3 Optimized crystal structure parameters of LaNi4Al

�ɱ�3������֪, Alԭ����3gλҪ����2cλ��19.72��13.6��10-3eV, ��Szajek ��[6]�����23.3��13.6��10-3eV����һ��, �ɼ�Alԭ��ȡ��LaNi5�����е�Niԭ�Ӳ����������, ��LaNi4Al(3g)�Ż���ľ��������ʵ���������, ��LaNi4Al(2c)�����ϴ�, �ɴ˿�����ΪAl��LaNi4Al��������ȡ��3gλ��Ni[6, 7]�� �����ں����LaNi4.5Al0.5�ļ����н�ֻ��Alȡ��3gλNiԭ�ӵľ���ģ�ͽ��м��㡣 �Ӽ����������Է���, Alԭ��ȡ��Niԭ�Ӻ�ʹ����������, ��c�����������a���Ҫ��, ʹ��a�� c��ֵ������С, ��������[4]ʵ����һ�¡� Alȡ��3g��λ��Ni��, ��Ӧ���м�һ���Ni��Al��λ����λ�ò�û�з����仯, ��λ��״̬�ı仯�����ڶ���͵���(basal plane)�� Alʹ�þ����������, ��ԭ�����ΪAl��ԭ�Ӱ뾶��Ni��ԭ�Ӱ뾶��, ��ԭ��ȡ��Сԭ�������˾���ij���, ����������; ��ʹ������λ��Niԭ���ܵ�3g��λ��Al�ļ�ѹ��
2.3 LaNi4.5Al0.5�Ľṹ����
�ڻ��Al����ɼ�λ�ú�, ���������״ζ�Alռ��Ϊ��������������˼��㡣 �ڹ�����������, �ڲ�����ԭ�ӿ������ڸ�λ�Գ��Եĸı�, ���зǶԳƵ�����, ����ڶԾ��������Ż���ͬʱ��Ҫ���ڲ�ԭ�Ӳ������г�ԥ, ���ܵõ������ϵ��Ż��ṹ�� ������ڱ�4��
��4 LaNi4.5Al0.5��ԭ����״̬���Ż�������
Table 4 Original and optimized structures of LaNi4.5Al0.5
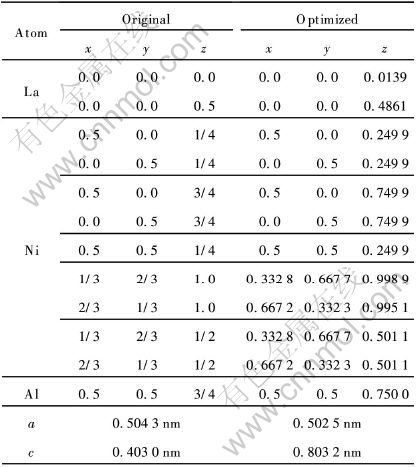
��LaNi4.5Al0.5�Ľṹ�������, �Ż������ʵ������Ϊһ��, ��a��c���������ԼΪ0.35%�� ϸ�µıȽϻ����Է���, Al�IJ��ӻ����¾���ṹ�IJ���������, ������Al�Ǵ���(0, 0.5, 0.75), ��˵��������ڽ�������La-La�ļ���������Ϊ0.4239nm, ��˶�Ӧ����������La-La����ܵ���ѹ������, ����ֵΪ0.3792nm��
�������и�ԭ����״̬�ı仯��LaNi4Al�еı仯���������ͬ, ��Alȡ��3g��λ��Ni��, ��Ӧ�ġ��м䡱���Ni��Al��λ����λ�ò�û�з����仯, ��λ��״̬�ı仯�����ڡ����桱�͡����桱(basal plane)�� ���������ܵ�3g��λ��Al�ļ�ѹ, ����ԭ����z���Ͼ��в�ͬ�̶ȵ�λ�ơ�
2.4 �ܴ��ṹ��״̬�ܶȶԱ�
�ܴ��ṹ�Լ���������Ƶ�״̬�ܶȶ��ڷ��������е�ԭ�ӳɼ������ϵ����Ե�����Ҫ�����塣 �ܼ���״̬�ܶȷ�ӳ�˵�λ��������ڵĵ��ӿ��ܵ�״̬���� �������߲���GGA Pedew96����������������Ż��ļ��νṹ, ͬʱ�������˺Ͻ��ܴ��ṹ�� �����ܼ��Լ�ͶӰ̬�ܶȵ�Ӱ�졣
2.4.1 LaNi5���ܴ��ṹ��̬�ܶ�
ͼ3��ʾΪLaNi5���ܴ��ṹͼ, ������Ef=-10.591eVΪ0�㡣 �Ƚ�ͼ4(a)��(d)��֪, ��LaNi5��Ef�����ĵ�����, Niԭ�ӵ�3d�����ͶӰ̬�ܶ�ռ�о���, ����Hector��[11]�Ľ��һ�¡� ��ͼ3(b)��֪, Laԭ�ӵ�4f�����DOS��Ҫ�����ڴ���EfԼ3.7eV��, ������[12]��Լ0.6eV, ��������[13]��Լ1.2eV�� Niԭ�ӵ�3d�����������ԼΪ3.7eV, ���������߷���[14]��X�����������[15]����һ�¡� �ɼ�, LaNi5�Ľ�������Ҫ��Դ��Ni�ĵ��ӵĹ��л��˶�, ��Ni�ĵ�����Դ��Ҫ��3d���, ��ͼ4(d)��ʾ; La�Ĺ�����Ҫ��4f����γɵļ۴���5p�ڲ�����(15.5eV), ��ͼ4(b)��ʾ��
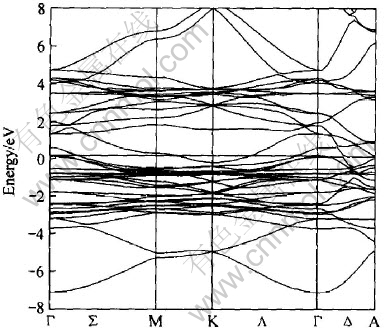
ͼ3 LaNi5���ܴ�ͼ
Fig.3 Energy band structure of LaNi5
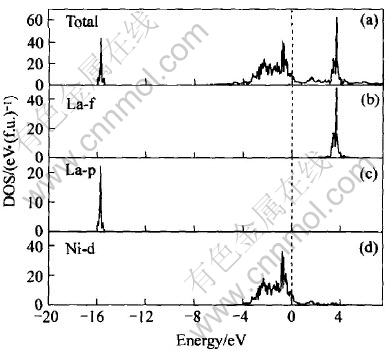
ͼ4 LaNi5�ܵ�DOS�Ͳ���DOS
Fig.4 Total and partial DOS of LaNi5
[TS)] ��(c)�� ��ͼ4(a)��, ��Ef����̬�ܶ�n(Ef)=10.1(11.81)eV/f.u.�ݴ����������Ϊ23.7(27.9)mJ/mol, �����ڵ����µ�ʵ��ֵ36.5mJ/mol[16], ������Hector��[11]�ļ���ֵ21.7mJ/mol��
2.4.2 LaNi4.5Al0.5���ܴ��ṹ��̬�ܶ�
���ڴ�ǰ��û�й���LaNi4.5Al0.5�ľ�����ӽṹ�о�������, ��˱������߸����˱Ƚ��������ܴ��ṹͼ(ͼ5)�������Լ�����ԭ�ӵķ���̬�ܶ�ͼ(ͼ6~9)�� ����ע�����, ����ʹ��˫�������ij�����, �������̬�ܶȺ�����̬�ܶ�Ӧ���ǵ����������� �Ƚ�LaNi4.5Al0.5��LaNi5��̬�ܶ�, ���Է������ߵ�������״���ɷ���������ͬ, ��Ni��3d����ռ���˵����ľ���Ȩ��, ��La��f������Ҫ�����˼۴�, La��5p���ӹ�����15.5eV������, Ni��s�� p�� f��������һ�����Ĺ��ײ��ɺ��ӡ� ����ͬ������LaNi4.5Al0.5��Al��s��p������̬�ܶȵĸ���Ҫ�������־��в�ͬ�̶ȵĹ���, ���ܷdz�С�� ����, �������˷�������������, Ef=-10.134eV, ����Ҫ����ΪAl��s��p�ṩ3���۵�������Niԭ�ӵ�10�۵�����, ��˵��²���Ԩ������ܼ�(�����ܼ�)�½��� ��ͼ5��Ҳ����ֱ�Ӹ����������̬�ܶ�Ϊ8.86eV/f.u., �ݴ˼������������Ϊ20.9mJ/mol��
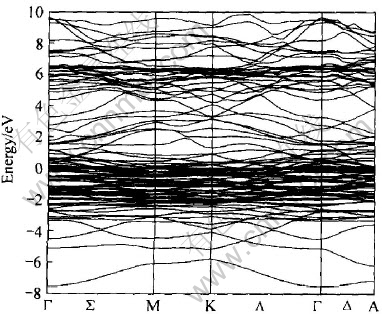
ͼ5 LaNi4.5Al0.5���ܴ��ṹ
Fig.5 Energy band structure of LaNi4.5Al0.5
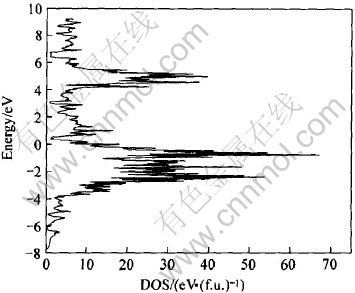
ͼ6 LaNi4.5Al0.5������̬�ܶ�
Fig.6 Total DOS of LaNi4.5Al0.5
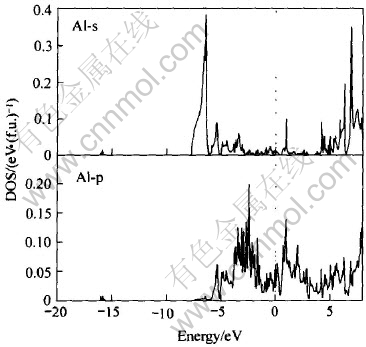
ͼ7 Al�ĸ�����̬�ܶ�
Fig.7 Partial DOS of Al
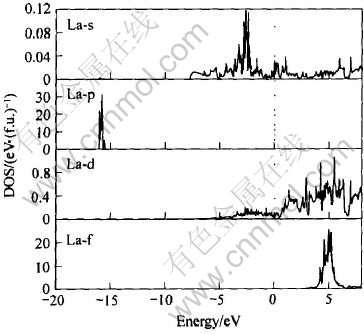
ͼ8 La�ĸ�����̬�ܶ�
Fig.8 Partial DOS of La
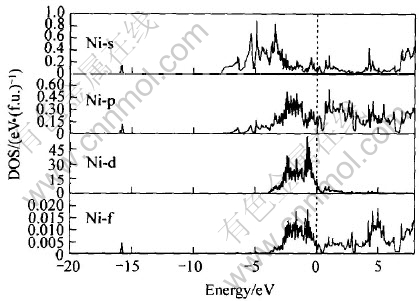
ͼ9 Ni�ĸ�����̬�ܶ�
Fig.9 Partial DOS of Ni
2.4.3 LaNi4Al���ܴ��ṹ��̬�ܶ�
ͼ10��ʾΪLaNi4Al���ܴ��ṹͼ, ������Ef=-9.441eVΪ0��, ��LaNi5�� LaNi4.5Al0.5����ķ����ܼ���, ��Ҳ������ƺ̨ѹ�½�����Ҫԭ�� ͼ11�� 12�� 13��ʾ�Ǹ�����Ӧ���ܴ��ṹ���Ƶ�̬�ܶ�ͼ�� ��ͼ11��֪, LaNi4Al�ļ۴�����ԼΪ8.2eV, ��Szajek ��[6]�����8.6eV����һ��; �ڷ����ܼ���n(Ef)=1.61; ��EF���ĵ�����Ҫ����La��4f����� Ni��3d�����Al��3s��3p������, ��ͼ12�� 13��ʾ��
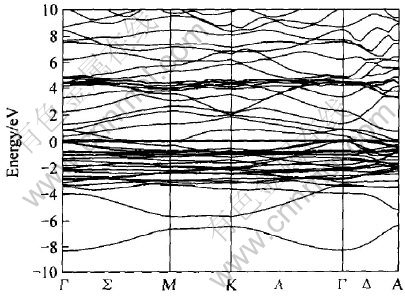
ͼ10 LaNi4Al���ܴ��ṹ
Fig.10 Energy band structure of LaNi4Al
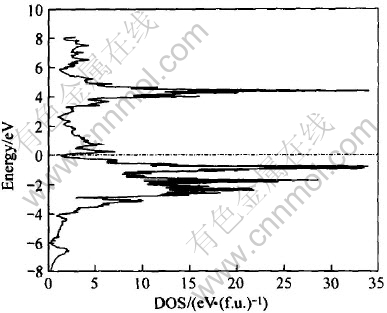
ͼ11 LaNi4Al������̬�ܶ�
Fig.11 Total of DOS for LaNi4Al
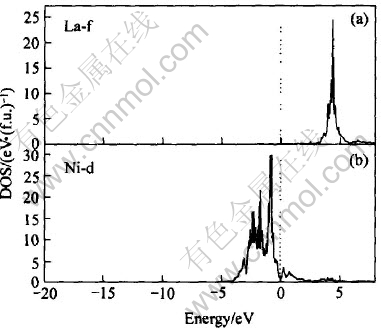
ͼ12 La��Ni�IJ���̬�ܶ�
Fig.12 Partial DOS of La and Ni
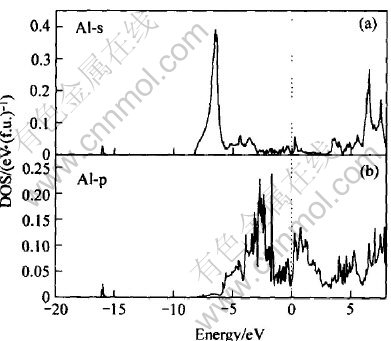
ͼ13 Al�ĸ�����̬�ܶ�
Fig.13 Partial DOS of Al
�Ƚ�ͼ11��ͼ12(b)��֪, ��LaNi4Al�ļ۴���, Niԭ��3d�����ͶӰ̬�ܶ�ռ��, n(Ef)=0.92, Ҳ��Szajek ��[6]��ͬ�� ��ͼ12(a)��֪Laԭ��4f�����ͶӰ̬�ܶ���Ҫ�����ڴ���EF(Լ4.382eV)��, ��Hector��[11]�����LaNi5����Զ������ܼ��� ��ͼ13��֪�ڷ����ܼ���, Alԭ�ӵ�3s�����3p����Գɼ�����һ���Ĺ���, ��3p����Դ���3s�����
��ͼ11�з����ܼ���̬�ܶ�(n(Ef)=1.61eV/f.u.)�ɵõ��ӱ�����ԼΪ3.7975mJ/mol, ���Ե���Hector��[11]�����LaNi5�е��ӱ�����23.7mJ/mol�� ����Alԭ�ӵļ���ʹ����������ȷ����仯, �Ӷ�Ҳ����ʹLaNi4Al�����仯������ȶ��Է���һ���仯��
ͨ����������������Է���, LaNi5-xAlx(x=0.0, 0.5, 1.0)�������� ������������ �������̬�ܶȼ����Ӧ�ĵ��±����ݵȲ�����Al�����ı仯�dz�����, �ȽϽ�����ڱ�5��
��5 Al������Ef, n(Ef)�ͦõ�Ӱ��
Table 5 Effects of Al content on Ef, n(Ef), ��
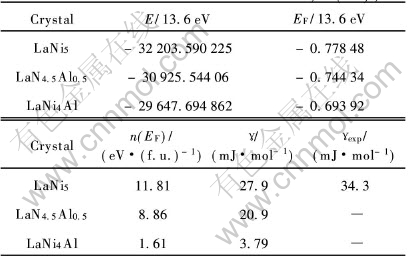
�ɼ�, ����Al����������, ���¾��������� ����������������, ��Al��0.0, 0.5���ߵ�1.0��, ������������-10.591eV�� -10.134eV���ߵ�-9.441eV, ��Ef�ϵ�̬�ܶȺͲ��϶�Ӧ�ĵ��±��������½�, ����Ҫ������Alԭ�ӵ�s��pֻ�ṩ��3���۵���, ����Niԭ�ӵ�10���۵������¡� ����LaNi5, ʵ��۲�ı�����Ϊ34.3mJ/mol, ����ֵ����ʵ��ֵ, ���Ǹ���Hector��[11]�����LaNi5�е��ӱ�����ֵ(23.7mJ/mol)�� ��LaN4.5Al0.5�� LaNi4AlĿǰ��û��ʵ���������ֵ, ��ʵ�����״θ�������Ӧ������ֵ��
3 ����
1) ��ȫ����ˮƽ��, ���ڹ����ݶȽ��ƺ�ȫ����������ƽ�沨����, ���������LaNi4Al����������� �ܴ��ṹ�� �����ܶȺ�״̬�ܶȡ�
2) ����LaNi5, ����GGA Perdew96��LSDA���ּ��㷽�������˼���, �������GGA Perdew96 �����ľ���ռ�ṹ��ʵ��ṹ���ݷ��ϵķdz��á� �������������Ż��ṹ��Ӱ�첻���ԡ�
3) ����LaNi4Al, ��������ģ�ͼ����������Ϊ, Alԭ��ֻ��ȡ��3g��λ��Niԭ�ӡ�
4) ��LaN4.5Al0.5�Ľṹ���������ۼ���, ����õ��ļ��νṹ������ʵ��ֵһ��, ���㻹�������������ܴ��ṹ��̬�ܶ��Լ����ԭ�ӵĸ����Ƕ���������ͶӰ̬�ܶȡ�
5) ����Al����������, ���������� ��������������, ���LaNi5, LaN4.5Al0.5��LaNi4Al, ������������-10.591�� -10.134���ߵ�-9.441eV, ��Ef�ϵ�̬�ܶȴ�11.81, 8.86���罵�͵���1.61eV/f.u.��
REFERENCES
[1]Dayan D, Mintz M H. Hysteris effects in cerium-containing LaNi5-type compounds [J]. Less-Common Met, 1980, 73: 15-24.
[2]Osumi Y, Suzuki H, Kato A, et al. Development of mischmetal-nickel and titanium-cobalt hydrides for hydrogen storage [J]. Less-Common Met, 1980, 74: 271-277.
[3]Sakai T, Oguro K, Miyamura H, et al. Some factors effecting the cycle lives of LaNi5 hydrogen batteries [J]. Less-Common Met, 1990, 161: 193-202.
[4]�ܴ���, ����, �亣��, ��. Al���Ni��LaNi5-xAlx���ܵ�Ӱ��[J]. �й�ϡ��, 2004, 25: 5-8.
CAO Da-li, ZHANG Rui-jing, LENG Hai-yan, et al. Effects of Al substitution for Ni on the properties of LaNi5-xAlx [J]. Chinese Rare Earths, 2004, 25: 5-8.
[5]���帻, �»���, ����. AlЧӦ��LaNi5-xAlxϵ�Ͻ��������ܵ�Ӱ��[J ]. ϡ�н��������빤��, 2005, 34(2): 240-243.
XIONG Yi-fu, CHEN Hu-chi, LUO De-li. Influence of aluminium-effect on absorption/desorption function of LaNi5-xAlx alloys [J]. Rare Metal Materials and Engineering, 2005, 34(2): 240-243.
[6]Szajek A, Jurezyk M, Rajewski W. The electronic and electrochemical properties of the LaNi5, LaNi4Al and LaNi3CoAl systems [J]. Alloys and Compounds, 2000, 307: 290-296.
[7]Percheron-Guegan A, Lartigue C, Achard J C. Neutron and X-ray diffraction profile analyses and structure of LaNi5, LaNi5-xAlx and LaNi5-xMnx intermetallics and their hydrides (deuterides) [J]. Less-Common Met, 1980, 74: 1-12.
[8]������, ������. LaNi4Al����Ͻ�ľ���ṹ[J]. ԭ���ܿ�ѧ����, 1998, 32: 456-460.
PENG Shu-min, ZHAO Peng-ji. Crystal structure of LaNi4Al alloy for storing hydrogen [J]. Atomic Energy Science and Technology, 1998, 32: 456-460.
[9]Perdew J P, Burke S, Ernzerhof M. Generalized gradient approximation made simple [J]. Phys Rev Let, 1996, 77: 3865-3868.
[10]Mukerjee S, McBreen J, Reilly J J, et al. In-situ X-ray absorption spectroscopy studies of metal hydride electrodes [J ] . Electromchem Soc, 1995, 142(7): 2278-2285.
[11]Hector L G Jr, Herbst J F, Capehart T W. Electronic structure calculations for LaNi5 and LaNi5H7: energetics and elastic properties [J]. Alloys and Compounds, 2003, 353: 74-85.
[12]Malik S K, Arlinghaus F J, Wallace W E. Calculation of the spin-polarized energy-band structure of LaNi5 and GdNi5[J]. Phys Rev B, 1982, 25: 6488-6491.
[13]Nakamura H, Nguyen-Manh D, Pettifor D G. Electronic structure and energetics of LaNi5, ��-La2Ni10H and ��-La2Ni10H14[J]. Alloys and Compounds, 1998, 281: 81-91.
[14]Weaver J H, Franciosi A, Wallace W E, et al. Electronic structure and surface oxidation of LaNi5, Er6Mn23, and related systems [J]. Appl Phys,1980, 51: 5847-5851.
[15]Fuggle J C, Hillebrecht F U, Zeller R, et al. Electronic structure of Ni and Pd alloys(��): X-ray photoelectron spectroscopy of the vallence bands [J ]. Phys Rev B, 1982, 27: 2145.
[16]Takeshita T, Dublon G, McMasters O D, et al. The Rare Earths in Modern Science and Technology[M]. New York: Plenum,1980. 563.
������Ŀ: ������Ȼ��ѧ-�й����������о�Ժ���ϻ���������Ŀ(10276027)
�ո�����: 2004-12-29; ������: 2005-04-24
�����: ����(1969-), ��, ������, ��ʿ.
ͨѶ����: ����; �绰: 028-85405234; E-mail: gthhl@sina.com
(�༭ ������)

