中国有色金属学报 2004,(08),1274-1280 DOI:10.19476/j.ysxb.1004.0609.2004.08.004
微合金化Al-4.0Cu-0.3Mg合金时效初期微结构演变的计算机模拟
陈志国 李世晨 刘祖耀 郑子樵
中南大学材料科学与工程学院,中南大学材料科学与工程学院,中南大学材料科学与工程学院,中南大学材料科学与工程学院 长沙410083 ,长沙410083 ,长沙410083 ,长沙410083
摘 要:
采用Monte Carlo方法模拟了时效初期Al 4.0Cu 0.3Mg (0.4Ag) (0.2Sc)合金的原子分布。研究结果表明:在时效过程中含微量钪的Al Cu Mg合金中镁原子逐步向钪原子周围偏聚,而铜原子并没有向钪原子周围聚集的倾向,时效初期出现了大量的Mg/Sc原子团簇及Mg/Sc/空位复合体;微量钪的存在促进了镁原子团簇化,但抑制了铜原子的团簇化;而含微量银的合金中镁原子向银原子周围偏聚的倾向比铜原子大得多,时效初期出现了大量的Ag/Mg原子团;"Sc/空位"机制是微量钪影响Al Cu Mg合金时效初期原子分布与形态的关键所在。
关键词:
MonteCarlo方法 ;铝合金 ;时效 ;
中图分类号: TG115
作者简介: 陈志国(1969),男,博士,副研究员.; 陈志国,博士;电话:07318830270;传真:07318836032;E mail:zgchen@mail.csu.edu.cn;
收稿日期: 2004-05-25
Computer simulation of microstructural evolution of microalloyed Al-4.0Cu-0.3Mg alloys
Abstract:
The Monte-Carlo method was applied to simulate the distribution of solutes in the microalloyed Al-(4.0Cu-0.3Mg alloys) during the initial ageing stage. The simulation results show that the distribution and morpholo-(gy of the clusters have )been modified by trace element additions, and it is found that one of the key factors governing this effect is Sc/vacancy mechanism. Small additions of Sc to Al-Cu-Mg alloys exhibit a strong tendency to form Mg/Sc cluster and Mg/Sc/vacancy complexes in the atom configurations. The presence of Sc stimulates the clustering of Mg, while depresses the clustering of Cu. A small addition of Ag to Al-Cu-Mg alloys exhibits a strong tendency to form Ag/Mg( cluster. )
Keyword:
Monte-Carlo method; aluminium alloys; ageing;
Received: 2004-05-25
微合金化一直是铝合金的研究热点
[1 ,2 ,3 ]
。 近些年来, 尽管对银、 钪等在铝合金中的微合金化作用与机理取得了相当大的进展
[4 ,5 ,6 ,7 ]
, 但总体来说, 定性研究多, 定量研究少; 对析出相长大阶段研究的多, 初期形核研究的少; 在组织结构层次描述的多, 在原子层次进行系统探索的少。 由于对铝合金微合金化机制缺乏直接的、 动态的原子图像和基于统计的量化数据做支撑, 无法区分表观的与本质的因素对相变过程的影响。 通过计算机模拟合金化元素对相变初期溶质原子分布形态的影响, 能够弥补实验观测手段的不足, 从而进一步深入理解微合金化元素对铝合金微观结构演变的影响与作用机理。 Monte-Carlo方法作为探索原子分布的一种十分有效的方法
[8 ,9 ,10 ]
, 已成功应用于模拟有序相的相转变, 晶粒长大, 及铁磁相转变等方面
[11 ,12 ,13 ,14 ]
。 本文采用Monte-Carlo方法模拟了Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc)合金时效初期各溶质原子的分布状况, 并对银、 钪等微合金化元素影响合金时效初期微结构的演变机理进行了分析。
1 计算机模拟方法
1.1 建模
通过Monte-Carlo方法模拟合金中空位的随机游走过程而获得时效初期合金溶质原子的分布图。 为了便于建模, 使模拟过程在计算机上得以顺利实现, 对所研究的合金系统进行了如下理想化处理: ① 忽略了原子及原子簇尺寸因素对扩散过程的影响(即忽略晶格畸变对原子偏聚的影响); ② 原子间的相互作用只计最近邻原子的相互作用; ③ 忽略了空位与空位之间的相互作用; ④ 忽略了位错、 层错、 晶界等其他缺陷对扩散过程的影响; ⑤ 采用周期性边界条件; ⑥ 采用了纯铝在淬火温度下的平衡空位浓度作为时效的空位浓度(忽略了空位的湮没和崩塌成位错环的过程)。
空位迁移概率(w )取决于空位在迁移方向上最近邻原子交换位置前后的能量差异, 为了反映dE 如何影响空位的随机游走过程, 这里采用了Symmetrical Solution作为空位迁移的判据
w
=
exp
(
-
d
E
/
(
k
Τ
)
)
1
+
exp
(
-
d
E
/
(
k
Τ
)
)
?
?
?
(
1
)
式中 dE 表示空位迁移而引起的能量变化; k 是Boltzman常数。
由于这里仅考虑最近邻原子的相互作用, 因而dE 就等于交换位置后空位和交换原子周围形成最近邻原子对的能量和减去交换前它们的能量和, 即
dE =E after -E before
=(∑ε v (j )i ε j (v )i ′
(∑ε v (v )i ε j (j )i ′
式中 ∑ε v (j )i j 原子本身位置处与最近邻原子形成空位原子对的能量和; ∑ε j (v )i ′j 原子在空位本身位置处与最近邻原子形成原子对的能量和; ∑ε v (v )i ε j (j )i ′j 原子在原子本身位置处与最近邻原子形成原子对的能量和。
1.2 原子相互作用参数的选定
原子相互作用能从已知的热力学或动力学数据
[15 ,16 ,17 ]
中推导出来, 具体推导方法如下。
根据Sanchez等
[18 ]
的研究, 同种原子对之间的相互作用势可表示为
ε ii ε
i
i
Ρ
u
r
e
[(r
i
i
Ρ
u
r
e
/r )8 -2(r
i
i
Ρ
u
r
e
/r )4 ] (3)
式中 r
i
i
Ρ
u
r
e
是纯金属中原子间的最近邻距离, 即晶格常数; r 是原子势场的作用距离; ε
i
i
Ρ
u
r
e
指基于挥发能的原子之间的相互作用能。
ε
i
i
Ρ
u
r
e
=2Ecoh
式中 Ecoh
而异种原子对之间的相互作用能可由同种原子对之间的相互作用能数据修正而得出。
ε
i
j
=
1
2
(
ε
i
i
+
ε
i
j
)
+
Δ
ε
?
?
?
(
6
)
Δε 为修正项。
Δ
ε
=
R
Τ
j
max
[
ln
(
1
-
C
j
max
)
-
l
n
C
j
max
]
Ν
a
Ζ
(
1
-
2
C
j
max
)
?
?
?
(
7
)
在计算原子与空位的相互作用能参数时, 本文采用了Doyama等
[19 ]
的研究成果, 将原子与空位的相互作用能关系式表达
ε iv ε
i
v
Ρ
u
r
e
(r
i
i
Ρ
u
r
e
/r ) (8)
式中 ε
i
v
Ρ
u
r
e
=(E F V +E coh )/Z , 代表纯金属中原子与空位的相互作用能。
根据上述原理与算法, 采用Visual C++ 开发了基于Window平台的计算机模拟软件, 并对Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc)合金进行了模拟计算。
2 模拟条件
1) 模拟的合金化学成分为(质量分数, %): 合金A: Al-4.0Cu-0.3Mg; 合金B: Al-4.0Cu-0.3Mg-0.4Ag; 合金C: Al-4.0Cu-0.3Mg-0.2Sc; 合金D: Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc。
2) 模拟的时效温度为453 K(180 ℃)
3) Monte-Carlo循环次数8×108 次(对应于时效过程开始十几秒), 其间设置21个断点, 这21个断点分别为Monte-Carlo循环次数为2、 1×104 、 2×104 、 4×104 、 8×104 、 1×105 、 2×105 、 4×105 、 8×105 、 1×106 、 2×106 、 4×106 、 8×106 、 1×107 、 2×107 、 4×107 、 8×107 、 1×108 、 2×108 、 4×108 、 8×108 处, 以观察在453 K下时效初期各个不同时段溶质原子的分布变化情况。
3 模拟结果与分析
图1所示为合金Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc)时效初期铜、 镁原子簇的形态模拟结果。 从模拟结果可以发现, 在基合金Al-4.0Cu-0.3Mg中存在较大的空位团, 且在空位周围存在大量的镁原子及少量的铜原子。 而在添加了微量银的合金B中, 存在较多的Ag/Mg原子团。 与不含银的合金A比较起来, 合金B中空位团的尺寸小得多, 但这种空位团的数量较多, 这可能是由于微量银与镁的强烈交互作用从而降低了镁原子捕获空位的能力。 含微量钪的合金C中出现了大量的Mg/Sc原子团, 其中的空位几乎都被钪原子包围, 形成了大量的Sc/空位复合体, 与此同时也存在Mg/Sc/空位复合体。 在同时添加银与钪的合金D中出现了大量的Mg/Sc/空位复合体, 同时也存在少量的Ag/Mg原子团。
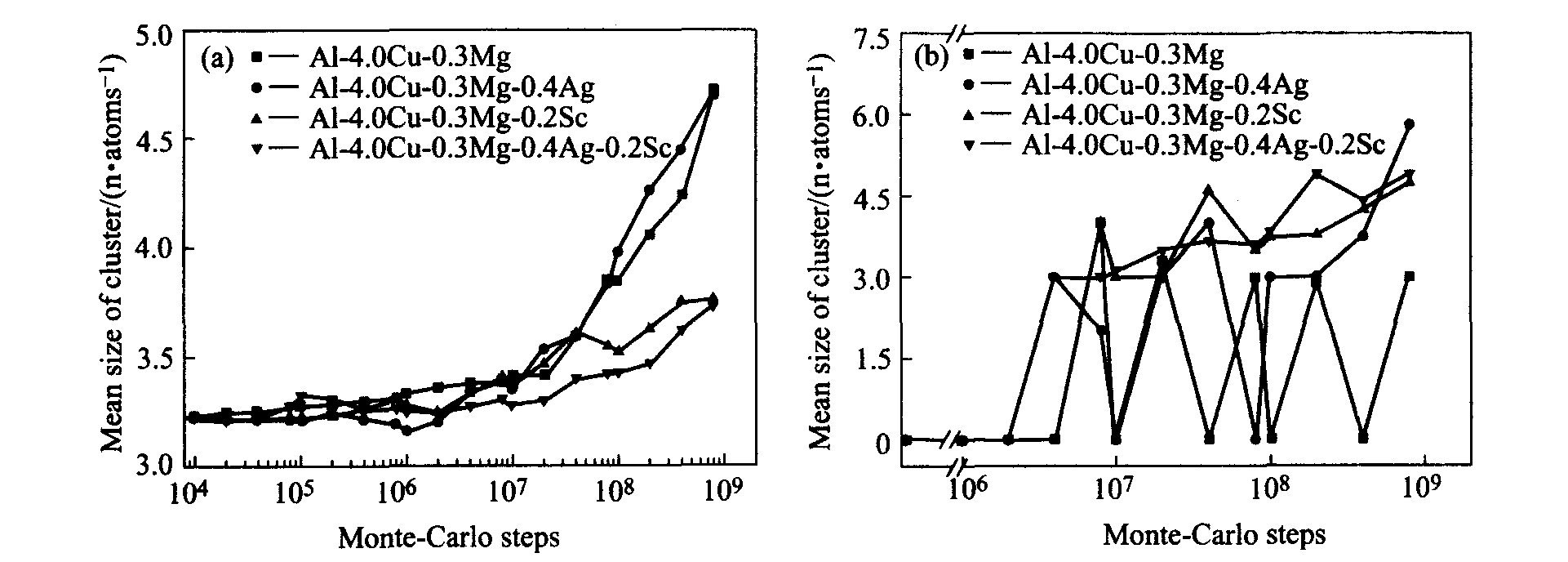
图2所示为模拟合金A、 B、 C、 D时效初期原子簇平均尺寸变化曲线。 从模拟结果上看, 微量钪的加入对铜原子的偏聚程度产生的影响较大。 从MCs=4×107 开始含微量钪的合金C与D中铜原子的偏聚程度明显地弱于不含钪的合金A、 B。 而相对钪来说, 微量银对铜原子偏聚的影响甚微。 从合金时效初期镁原子簇平均尺寸变化规律来看, 基合金A中的镁原子簇大小始终在同一水平上下波动, 但当添加微量银、 钪后, 其镁原子簇的平均尺寸随着时效时间的推移而稳步上升, 且其上升的起始时间顺序依次是: t D <t C <t B 。
图3所示为合金Al-4.0Cu-0.3Mg-0.4Ag-
图1 计算机模拟的Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc)合金实空间(001)α面溶质原子分布
Fig.1 Typical atom configuration from random (001)α 8 MCs (a)―Al-4.0Cu-0.3Mg; (b)―Al-4.0Cu-0.3Mg-0.4Ag; (c)―Al-4.0Cu-0.3Mg-0.2Sc; (d)―Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc
图2 Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc)合金时效初期原子簇的平均尺寸
Fig.2 Mean size of cluster in Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc) alloys during initial ageing stage (a)―Cu cluster; (b)―Mg cluster
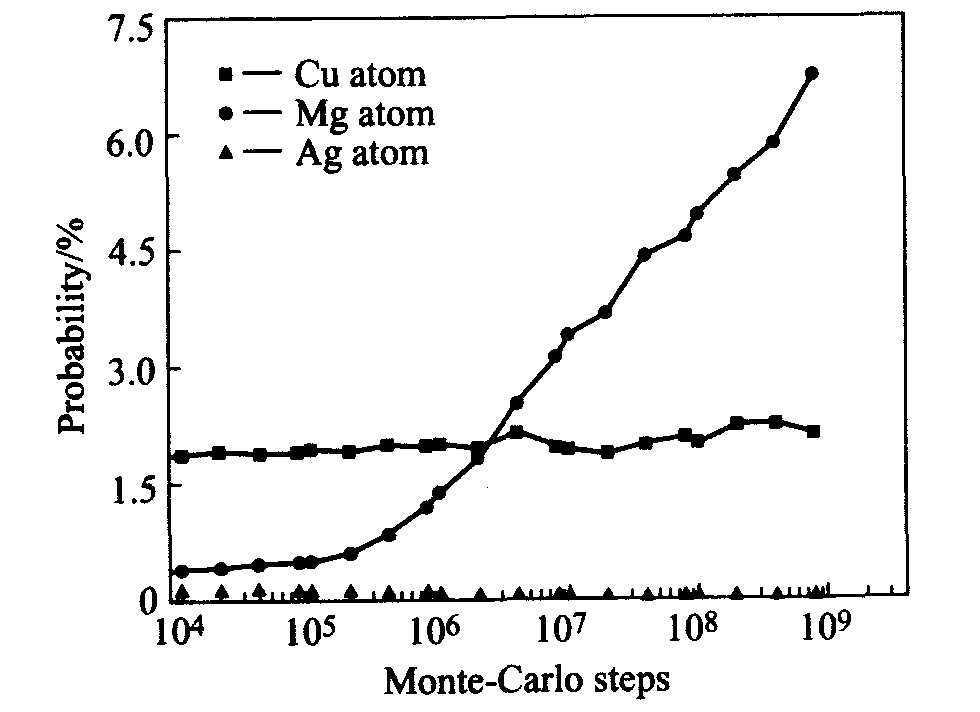
0.2Sc中钪原子周围出现铜、 镁及银原子的概率变化曲线。 从图中可以发现, 随着时效时间的推移, 钪原子周围出现镁原子的概率显著增加, 而钪原子周围出现铜原子与银原子的概率始终保持相对恒定。 这一结果表明在Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc合金中, 钪原子与镁原子之间表现出强烈的交互作用, 并具有很强的Sc/Mg原子团簇化倾向, 而钪原子与铜原子、 银原子之间并未表现出这种作用。
图3 Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc合金时效初期 钪原子最近邻位置出现其它溶质原子的概率变化
Fig.3 Probability of solute atoms positioned adjacent to Sc atoms in Al-4.0Cu-0.3Mg-0.4Ag- 0.2Sc alloy during early ageing stage
图4所示为合金Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc中银原子周围出现铜、 镁及钪原子的概率变化曲线。 由图可见, 银原子周围出现铜、 镁原子的概率均随着时效时间的延长而逐渐增加, 这说明在该合金中银原子与铜原子、 镁原子之间均存在一定的交互作用。 分析MCs=2与MCs=8×108 两者概率的增量值发现, 在银原子周围出现铜原子的概率仅从0.017增加到0.027(不到1倍), 而银原子周围出现镁原子的概率增加了7倍, 从而可以推测银原子与镁原子的交互作用比银原子与铜原子的交互作用大得多, 在该合金的时效初期具有强烈的Ag/Mg原子团簇化倾向。 最后, 对银原子周围出现钪原子的概率几乎没有变化甚至略有下降的分析认为, 银原子与钪原子之间并不表现出团簇化倾向。
图4 Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc合金时效初期 银原子最近邻位置出现其它溶质原子的概率变化
Fig.4 Probability of solute atoms positioned adjacent to Ag atoms in Al-4.0Cu-0.3Mg-0.4Ag- 0.2Sc alloy during early ageing stage
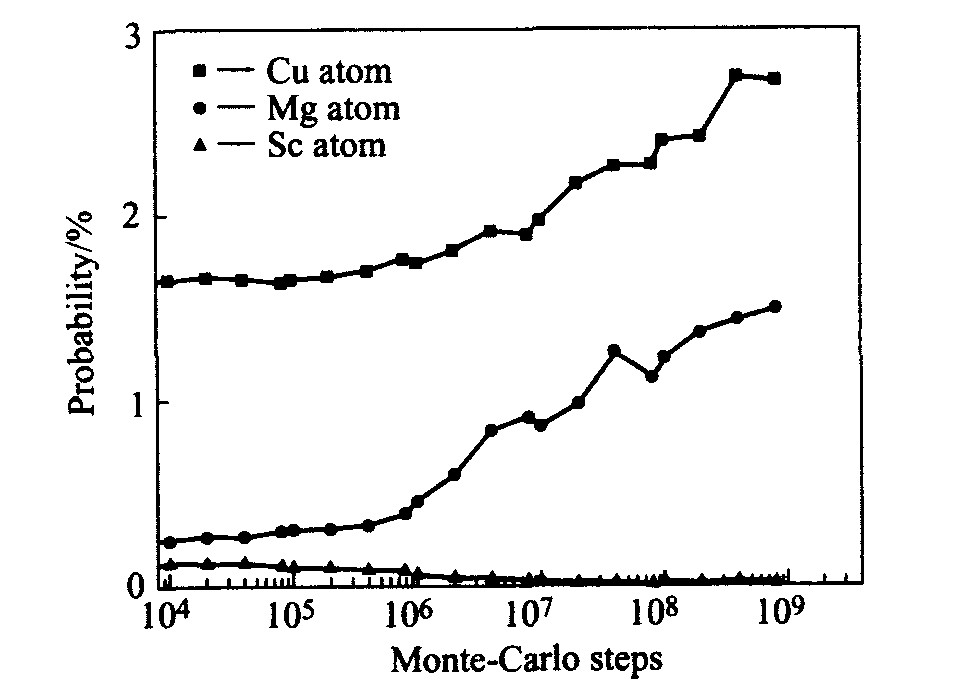
Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc)合金时效初期空位周围出现溶质原子的概率见图5。 由图可见, 随着时效时间的推移, 镁、 钪原子逐步向空位聚集, 但铜原子及所添加的银并不向空位处偏聚。 比较银、 钪微合金化前后4种合金中空位周围出现铜、 镁原子的概率发现(见图5(b), (c)), 不论钪还是银均不影响合金中空位周围的铜原子聚集状况。 然而空位周围的镁原子分布状况受到微量银、 钪的显著影响, 在基合金Al-4.0Cu-0.3Mg中添加微量钪使得合金中镁原子较未加钪的合金更显著地向空位处偏聚。 比较Al-4.0Cu-0.3Mg与Al-4.0Cu-0.3Mg-0.4Ag合金中空位周围镁原子的聚集状况, 发现在时效较早阶段含银合金中镁原子向空位偏聚的倾向较弱, 但当MCs>108 后, 其聚集程度十分强烈。 Al-4.0Cu-0.3Mg-0.2Sc与Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc合金中镁原子的聚集状况也出现了类似的情形。
4 讨论
综合以上模拟结果发现, 添加银、 钪等微合金化元素的Al-Cu-Mg合金在时效初期形核的关键阶段, 其中铜原子、 镁原子的偏聚程度和形态分布与未添加微量元素的基合金相比存在着显著的差异。 在时效初期微合金化铝合金中原子的偏聚程度和形态分布差异, 可首先从微合金化元素与溶质原子的交互作用上得到解释。 在所研究的合金中, 正是由于钪原子与镁原子、 银原子与镁原子之间存在强烈的交互作用, 因而在合金时效初期出现了Mg/Sc、 Ag/Mg原子团簇, 并使得其中镁原子团簇化倾向十分强烈。 从对Al-4Cu-0.3Mg系列合金时效初期溶质原子分布的模拟结果可知, 微量钪的添加促进了镁原子团簇化。
其次, 微量元素在合金相变过程中的作用往往与空位的相互作用密切相关。 从微量元素与溶质原子的交互作用分析可知, 钪原子与铜原子的交互作用较小, 然而在从对Al-4Cu-0.3Mg系列合金时效初期溶质原子分布的模拟结果可知, 微量钪的添加抑制了其中铜原子的团簇化, 这要从微量元素与其中空位的交互作用得到解释。 正是由于钪原子与空位之间存在较强的相互吸引作用, 使得合金中空位的可动性降低, 铜原子赖以扩散的空位急剧减少, 从而使得Al-Cu-Mg系合金中的铜原子簇丛聚程度大幅度降低。 为了直观表述钪在铝合金中的这种
图5 Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc)合金时效初期 空位周围出现溶质原子的概率变化
Fig.5 Probability of solute atoms positioned adjacent to vacancy in Al-4.0Cu-0.3Mg-(0.4Ag)- (0.2Sc) alloys during early ageing stage (a)―Solute atoms in Al-4.0Cu-0.3Mg-0.4Ag-0.2Sc; (b)―Cu atom in Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc); (c)―Mg atoms in Al-4.0Cu-0.3Mg-(0.4Ag)-(0.2Sc) alloys
作用机制, 这里称之为“Sc/空位”机制。
此外, 由于溶质原子与铝原子尺寸的差别, 原子团的形成势必在基体周围形成一定的应变场, 其中某些原子团的形成产生拉应变场, 另一些原子团的形成会产生压应变场。 当原子半径比铝原子半径大的溶质原子吸附在拉应变区时其应变能会降低, 同样地, 体应变能会在压应变区域吸附比铝原子半径小的原子或空位而降低。 在Al-4.0Cu-0.3M-0.4Ag合金中, 如果仅从对铜原子簇的聚集程度和分布的影响上看, 微量银的添加作用甚微。 但由于镁、 银之间的交互作用使得时效初期出现了大量Mg-Ag原子对, 从原子尺寸因素考虑, 镁原子半径比铝原子大得多, 而银原子半径与铝原子差不多, 正如镁原子在(111)α
[20 ]
, 出于降低应变能的需要Mg-Ag原子对也会在{111}α α α Ω 相的形核部位, 使铜原子簇易于在该部位偏聚, 从而促进了Ω 相形核析出。 以上原子尺度上微合金化元素作用机理的认识很好地解释了Murayama等
[21 ]
利用三维原子探针(3DAP)对Al-1.9Cu-0.3Mg-0.2Ag(摩尔分数, %)合金进行微观组织结构分析的结果, 同时使Ringer等
[22 ]
对含微量银Al-Cu-Mg合金时效早期就开始析出Ω 相的电镜观察结果得到深刻理解。
5 结论
1) 在Al-Cu-Mg合金中添加微量钪, 时效初期出现了大量的Mg/Sc原子团簇及Mg/Sc/空位复合体。 随着时效时间的推移, 镁原子逐步向钪原子周围偏聚, 而铜原子没有向钪原子周围聚集的倾向。 微量钪的存在促进了镁原子团簇化, 但抑制了铜原子的团簇化。
2) 在Al-Cu-Mg合金中添加的微量银, 合金中的镁原子向银原子周围偏聚的倾向比铜原子大得多, 时效初期出现了大量的Ag/Mg原子团簇。
3) Sc/空位机制是微量钪影响Al-Cu-Mg合金时效初期原子分布与形态的关键所在。
参考文献
[1] PolmearIJ.Controlofprecipitationprocessesandpropertiesinagedaluminiumalloysbymicroalloying[J].MaterialsForum,1999,23:117135.
[2] RingerSP,RaviprasadK.Developmentsinage hardenablealuminiumalloysandrationaldesignofmicrostructure[J].MaterialsForum,2000,24:5994.
[3] PolmearIJ.RoleoftraceelementsinagedAluminiumalloys[J].MaterialsScienceForum,1987,13/14:195214.
[4] RingerSP,HonoK.Microstructuralevolutionandagehardeninginaluminiumalloys:atomprobefield ionmicroscopyandtransmissionelectronmicroscopystudies[J].MaterialsCharacterization,2000,44:101131.
[5] HoweJM.AnalyticaltransmissionelectronmicroscopyanalysisofAgandMgsegregationto{111}θprecipitateplatesinanAl Cu Mg Agalloy[J].PhilosophicalMagazineLetters,1994,70(3):111120.
[6] MukhopadhyayAK.NucleationofΩphaseinanAl CuMgalloycontainingsmalladditionsofAg[J].MaterialsTransactions,JIM,1997,38(5):478482.
[7] CHENZhi guo,ZHENGZi qiao.MicrostructuralevolutionandageingbehaviourofthelowCu:MgratioAl CuMgalloyscontainingscandiumandlithium[J].ScriptaMaterialia,2004,50(7):10671071.
[8] HirosawaS,SatoT,YokotaJ,etal.ComparisonbetweenresisivitychangesandMonte CarlosimulationforGPzoneformationinAl Cubaseternaryalloys[J].MaterialsTransaction,JIM,1998,39(1):139146.
[9] HirosawaS,SatoT,KamioA.ClassificationoftheroleofmicroalloyingelementsinphasedecompositionofAlbasedalloys[J].ActaMater,2000,48(8):17971806.
[10] BinkeleP,SchmauderS.AnatomisticMonte Carlosimulationofprecipitationinabinarysystem[J].ZMetallkd,2003,94(8):858863.
[11] GaoJ,ThompsonRG.Realtime temperaturemodelsforMonte Carlosimulationsofnormalgraingrowth[J].ActaMater,1996,44(11):45654570.
[12] SaitoY,HaradaH.TheMonte CarlosimulationoforderingkineticsinNi basesuperalloys[J].MaterialsScienceandEngineeringA,1997,223A(12):19.
[13] PouduriR,ChenLQ.Computersimulationofthekineticsoforder disorderandphaseseparationduringprecipitationofδ′(Al3Li)inAl Lialloys[J].ActaMater,1997,45(1):245255.
[14] DeurinckP,CreemersC.Monte CarlosimulationofCusegregationandorderingatthe(110)surfaceofCu75Pd25[J].SurfaceScience,1998,419:6277.
[15] KittelC.IntroductiontoSolidStatePhysics[M].NewYork:JohnWiley&SonsInc,1976.57142.
[16] LandoltB.NumericalDataandFunctionalRelationshipinScienceandTechnology(Vol.25):AtomicDefectsinMetals[M].Berlin:Springer Verlag,1991.35117.
[17] LandoltB.NumericalDataandFunctionalRelationship
inScienceandTechnology(Vol.5):PhaseEquilibra,Crystallographic andThermodynamicDataofBinary[M ].Berlin:SpringerVerlag,1991.26129.
[18] SanchezJM ,BarefootJR ,JarrettRN ,etal.Model ingofγ/γ′phaseequilibriuminthenickel aluminumsystem[J].ActaMetallurgica,1984,32(9):15191525.
[19] DoyamaM ,KoehlerJS .Therelationbetweenthefor mationenergyofavacancyandthenearestneighborin teractionsinpuremetalsandliquidmetals[J].ActaMetallurgica,1976,24(9):871879.
[20] SuhIS ,ParkJK .InfluenceoftheelasticstrainenergyonthenucleationofΩphaseinAlCuMg(Ag)alloys[J].ScriptaMetallMater,1995,33:205211.
[21] MurayamaM ,HonoK .Threedimensionalatom probeanalysisofprecipitateclusteringinanAlCuMgAgal loy[J].ScriptaMater,1998,38(8):13151319.
[22] RingerSP ,HonoK ,PolmearIJ ,etal.NucleationofprecipitatesinagedAlCuMg(Ag)alloyswithhighCu∶Mgratios[J].ActaMater,1996,44(5):18831898.