����ԭλ�ۺϾ۰��������Ի�����֬
�����٣�¬ ��
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ������ ��ɳ��410083)
ժ Ҫ���Խ龧����4, 4��-��(��-��������)����(���DHBP)Ϊԭ�ϣ�ͨ������龧����(DHBP)���ƻ������ܣ�����ԭλ�ۺϾ۰����ķ������õ��ۺ��������õľ۰��������ԵĻ�����֬������ϵ���ϲ��ϡ�ʵ������������DHBP����Ϊ������֬E51��6%����TDIĦ����Ϊ1?2��DDM����Ϊ35%����Ӧʱ��Ϊ2.5 h�������£����Ʊ����ϵij��ǿ�������187.9%������ǿ�����4.7%������ǿ�����19.5%���ȷֽ��¶ȺͲ������¶Ⱦ�����Լ9 �档�����ͨ��ԭλ�ۺϵķ������Դ����߸��ϲ��ϵ����ԣ���ͬ�̶ȵظ��Ƹ��ϲ��������ۺ����ܡ�
�ؼ��ʣ�������֬��DHBP���۰�֬�����ͣ�ԭλ�ۺ�
��ͼ����ţ�TQ314.2��TQ322.4+1 ���ױ�ʶ�룺A ���±�ţ�1672-7207(2008)05-0993-07
Toughening modification of epoxy resin through
in-situ polymerization of polyurethane
DENG Xiao-qin, LU Bin
(School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: Polyurethane was made by in-situ polymerization process using mesogenic diol 4, 4��-di(��-hydroxethyl) xenene (DHBP) as raw materials in the matrix of epoxy resin. Due to the introduction of DHBP, the performance of parent metal was improved. Then, the toughening modified epoxy resin composites with excellent properties were prepared. The system could be seen as homogeneous composite material. The results indicate that when the content of DHBP is 6%, n(DHBP)/n(TDI)=1?2, dosage of DDM is 35%, reaction time is 2.5 h, the impact strength, bend strength and tensile strength of the modified epoxy resin composites are increased by 187.9%, 4.7% and 19.5%, respectively. The decomposing temperature and the glass transition temperature of the composites both increase by 9 ��. Thus through the in-situ polymerization, the overall performance of composite materials is greatly enhanced.
Key words: epoxy resin; DHBP; polyurethane; toughening; in-situ polymerization
������֬��һ���ۺ�����������ȹ��Ը߷��Ӳ��ϣ������ڽ����ȸ��������ʴࡢ�ͳ�����ܲ�[1-3]��������֬�����Է���������������൯������ ��[4-5]����չ��������������[6-7]��������Һ���߷�������[8]��������������[9-10]��������������[11-12]�Լ�ԭλ�ۺ�������[13-14]�ȷ��������У�������������Ի�����֬���ͷ�����ɴﵽ�Ϻõ�����Ŀ�ģ��������������뻷����֬�IJ��������ԣ����������ܽ��ڻ����У����������ϵ����ģ���벣�����¶��½���Ӱ�����Ч�������ù������ϸ��Ի�����֬���ͷ���Ҳͬ�������������⡣����������Һ���ۺ����۵�ߣ�����һ�㷽�����ѽ�����ȷ�ɢ�ڻ�����֬�����У����ѻ��Ԥ�ڵ�����Ч��[15]������������������һ����֮��Ч�������Է�������ʵ���д�������ν����ײ��Ͼ��ȷ�ɢ�ڻ�����֬�еĹؼ�����[16] �������ۺ�������(IPN)��һ�ֱȽ���������ͷ���[17]�����ø÷��������ܴﵽ���ͻ�����֬��Ŀ�ģ�ͬʱ���ܲ������俹���ǿ�ȺͿ���ǿ�ȡ�ԭλ�ۺϿ�ʹ����̬���Ը߷��Ӿ��ȷ�ɢ�ڸ��Ի����У��˷����������һ�������Ծۺ������������֬���Ե�ͬʱ��������ѧǿ�ȡ�ģ���������Ե�ȱ�㣬Ҳ�����Һ���ۺ�����Ѿ��ȷ�ɢ�ڻ�����֬�����ж�Ӱ��ʵ��������Ч��������[18]���Ӷ��ﵽ����ˮƽ�ϸ������͵�Ŀ�ġ�
���ڻ�����֬�ĸ��ԣ����ó��õķ������������Ե�ͬʱ���������˲��ϵ�ģ�����������ܵȡ������ý�Ϊǰ�ص�Һ���ۺ�����Ի�����֬���ͷ�����������������ȷ����ѧ���ܺ��������ܲ����ͣ����Dz��ø÷����ѶȺܴ������ɱ��ܸߡ��ڴˣ��������������ԭλ�ۺϵķ�������������û����ۺ������編���ص㣬���������ڻ�����֬��ԭλ�ۺϵĵ������������Ԥ����Ϊ�����㣬ͨ������龧�����������ƻ�����ڲ��ṹ��̽�����ڻ�����֬��ԭλ���ɴ���Ӻ������Ӽ���ɢ�ڻ�����֬�еĿ������Լ���Ӧ����������ԭλ�ۺϷ����Ʊ�����Ϊ������ϵ�ľ۰���/������֬���ϲ��ϡ�
1 ԭ���ϼ����鷽��
1.1 ��Ҫԭ���Ϻ��Լ�
������֬��E51������ʯ�ͻ����ܳ�������֬��������2, 4-�ױ�����������(TDI)����ѧ�����й�ҽҩ�����Ϻ���ѧ�Լ���˾�������������ӣ����ձ����ڣ�4, 4��-��������������(DDM)����ѧ�����Ϻ�����˾�Լ�����˾������2-���Ҵ�����ѧ�����й��Ϻ������Լ���������N, N-����������(DMF)�����������㶫��ͷ����¤������������
1.2 �м���4, 4��-��(��-��������)�����ĺϳ�
��Ħ����1?4ȡһ�����������Ӻ�2-���Ҵ���250 mL������ƿ�У�������ˮ�Ҵ�������NaOH�����������Ӧ24 h������Ӧ�ﵹ����ˮ�У����˵õ������Ȼ���������Ϊ3?1����ˮ�Ҵ���DMF����ܼ��ܽ⣬���˳�ȥ���ʣ������˵õ��ľ������ܼ��Ļ�������¼������ܽ⣬��ȴ�ᾧ3 h���ϣ������˵õ����壻�ڳ�����ո�����(ѹ��1 kPa)���T��4, 4��-��(��-��������)����(DHBP)��
1.3 ������ϵ��Ʊ�
��������ƿ�м��뻷����֬��������160~180 ���ˮ����Ȼ����DHBP���壬������140~150 �沢����ʹ���ܽ⣻������110 ������ʱ�����뻯ѧ������TDI��������������110 �����ң����跴Ӧ1 h������������80 �����ң������ۻ���DDM�����ȣ�80 ���������ע����Ԥ�Ⱥõĸ�ģ�У��ڱ��̻���������80 �汣��2 h����150 �汣��4 h����ȴ��ģ������й����ܡ��������ܵ�ⶨ�Dz�ý龧������ת���¶�Ϊ210 �档��ѧ��ӦʽΪ��
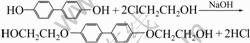 ��
��
2 ���������
2.1 ���������
ͼ1��ͼ2��ʾ�ֱ�ΪE51/TDI��E51/DHBP�Լ�E51/DHBP/TDI������ϵ��Ӧ1 h�ĺ�����ͼ����ͼ1��ͼ2���Կ�������915 cm-1���������ԵĻ�������ǿ��û�м�����˵�����������뻷�����ŷ�Ӧ�̶Ⱥ�С����ͼ2(b)���Կ�������1 735 cm-1�����������շ壬��Ϊ�۰����а�����������C=O�����շ壬���ڵ���DHBP��TDI�о������ڰ�������������(C=O)����˵��DHBP��TDI�����˷�Ӧ������2 266 cm-1���������������շ����Լ�������2 266 cm-1���������շ壬����Ϊ��ϵ�м����˹�����TDI������DHBP��TDIԭλ�����˶�����������Ԥ���塣��Ӧʽ���£�


ͼ1��E51/TDI������FTIRͼ
Fig.1 FTIR spectrum of E51/TDI mixture
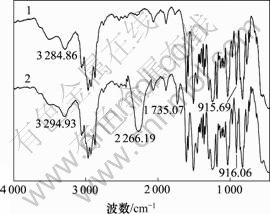
1��E51/DHBP; 2��E51/DHBP/TDI
ͼ2 E51/DHBP��E51/DHBP/TDI��ϵ��FTIRͼ
Fig.2 FTIR spectrum of E51/DHBP and E51/DHBP/TDI systems
2.2 �����;۰���/������֬������ϵ���ѧ����
DHBP�ɱ�ʾΪ��HO���龧��Ԫ��OH���龧��Ԫ�Ǹ����½���š�Ϊ��ʹ�����;۰����뻷����֬�нϺõ������ԣ�ѡ���뻷����֬�ṹ���������������Ϊ�龧��Ԫ����ṹʽ���£�
 (2)
(2)
��ʵ�鷢�֣���ֱ�Ӱ��������Ӽ��뵽������֬�У�����TDI��Ӧ1 h��Ȼ����̻���DDM���̻�����Ʒ����ѧ���ܺܲ���ǿ�ȱȴ�������֬�ij��ǿ�Ȼ�Ҫ�ͣ����������������ӵ��ǻ�(���ǻ�)��TDI�ķ�Ӧ����С��ԭλ��Ӧʱδ���γɶ���������Ԥ���塣��ˣ�ʵ���о�����Ԥ�Ⱥϳɴ��в��ǻ��Ľ龧�������仯ѧ��ӦʽΪ��
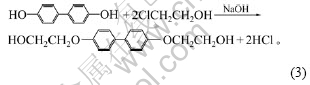
2.2.1 DHBP�����Թ��������ѧ���ܵ�Ӱ��
ͼ3��ʾΪE51�ͻ�����֬��ϵ�У�DHBP��TDIĦ����Ϊ1?2��DDM����Ϊ30%����Ӧʱ��Ϊ1 hʱ��������ϵ���ѧ������DHBP�����Ĺ�ϵ����ͼ3���Կ��������Ž龧�������������ӣ����ǿ�ȡ�����ǿ�Ⱥ�����ǿ�������ߣ�Խ����ֵ�������½������ǿ���ڽ龧��������Ϊ5%���ҳ������ֵ���봿E51��ȣ����ǿ�ȴ�ԭ����15.73 kJ/m2�����ӵ�44.17 kJ/m2�����Լ180.8%������ǿ����Խ����ֵ���½�����Ҳ��ԭ����91.16 MPa���ӵ�96.6 MPa���ң����Լ6.0%������ǿ�ȴ�59.5 MPa���ӵ�72.03 MPa�����Լ21.1%���ﵽ���ֵ�Ժ���ǿ�Ⱥ���ѧģ�����½������ƣ�������ΪDHBP���������Ӻ� ���Ӹ��ϳ̶Ƚ��ͣ��۰����뻷����֬������������ij̶ȼӴ����;۰����ڻ�����֬����������γ�Ӧ�����е㣬�Թ�����ϵ���ѧ��������Ӱ�죬��ʹ������ϵ���ѧ�����½���
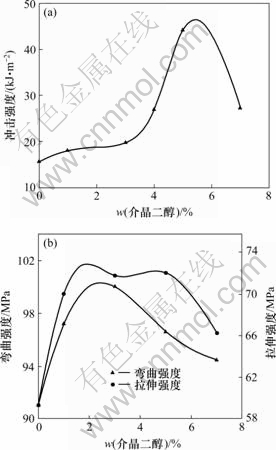
(a) ���ǿ��; (b) ����ǿ�Ⱥ�����ǿ��
ͼ3��DHBP�����Թ��������ѧ���ܵ�Ӱ��
Fig.3 Effects of DHBP content on mechanical properties of mixture
2.2.2 DDM�����Թ��������ѧ���ܵ�Ӱ��
ͼ4��ʾ��DHBP����Ϊ6%��DHBP��TDIĦ����Ϊ1?2����Ӧʱ��Ϊ1 hʱ��������ϵ���ѧ������DDM�����Ĺ�ϵ����ͼ4���Կ���������DDM���������ӣ����ǿ�ȡ�����ǿ�Ⱥ�����ǿ�ȶ��н����Ե���ߡ���DDM����Ϊ27%��ȣ�DDM����Ϊ35%ʱ�����ǿ�����185.4%������ǿ�����4.7%������ǿ�����19.5%��DDM��������33%ʱ������ǿ�Ⱥ�����ǿ�����ӷ�������ƽ����DDM��˫�����ã�һ���뻷�����ŷ�����Ӧ�̻�������֬�����dz䵱���������뻷����֬�����ɵĶ���������Ԥ���巴Ӧ���ڻ��������ɸ��Դ���ӡ�
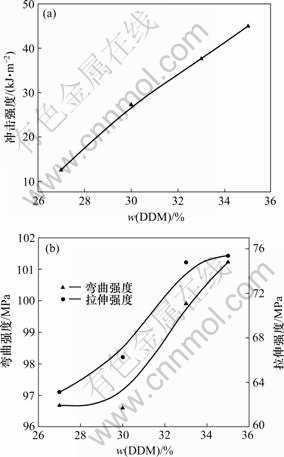
(a) ���ǿ��; (b) ����ǿ�Ⱥ�����ǿ��
ͼ4��DDM�����Թ��������ѧ���ܵ�Ӱ��
Fig.4 Effects of dosage of DDM on mechanical properties of mixture
����CYD128������֬���ο�������Ϊm(CYD128)?m(DDM)��100?28[19]������6%�Ľ龧���������̻�������Ϊ5%~7%ʱ���̻��������ȽϽӽ��ڶ��ǻ����������������ʹDHBP�������ɾ۰�������DDM�����ϵ�ʱ�����ɷ����;۰�����������Ӧ�̶Ƚϵͣ���������Ӧ�̶���ϵͣ����¹̻��サ���ܶȵͣ����γ��������״�ṹ[2]����ʹ������֬������ϵ���ѧ������Խϵ͡�����DDM����ʱ����ʹ������������Ԥ�����γɶ˰����ĺ������ε�Ԥ���壬�ڹ̻���Ӧʱ�뻷����֬�Ļ������ŷ�Ӧ������֦�ڻ�����֬�����У���֦�ľ۰���������֬�������Σ����ҽ龧��Ԫ�����Ѽ����������������֬�ij��ǿ�ȣ�ͬʱ����DDM����ʱ��������δ����ȫ��Ӧ��ʹ�л����������а������������˶�ʹ��״�ṹ���ɣ�δ���γ��������״�ṹ������ʹ������֬�ij��ǿ���в������������ƣ����ʹ������ǿ��������ǿ�ȱ仯��������ƽ����
2.2.3 DHBP��TDI��Ӧʱ��Թ��������ѧ���ܵ�Ӱ��
ͼ5��ʾ��DHBP����Ϊ6%��DHBP��TDIĦ����Ϊ1?2��DDM����Ϊ35%ʱ��������ϵ���ѧ�����뷴Ӧʱ��Ĺ�ϵ����ͼ5���Կ��������ŷ�Ӧʱ������ӣ�������ϵij��ǿ����һ������ߣ���Ӧ2.5 h����ǿ��Ϊ45.29 kJ/m2���ȷ�Ӧ1 h�ij��ǿ��37.55 kJ/m2���20.6%������ǿ���ڷ�Ӧ1 h��Ϊ101.05 kJ/m2����Ӧ2.5 h��Ϊ104.07 kJ/m2�����3.0%������ǿ���ڷ�Ӧ1 hʱΪ75.537 MPa����Ӧ2 hʱΪ77.941 MPa�����3.2%����Ӧʱ�䳬��2.5 h������ǿ�ȷ����½���������Ϊ���ŷ�Ӧʱ����ӳ���������������Ӧ�ij̶���ߡ�������ϵ���ѧ�������ŷ�Ӧʱ����ӳ��������߷�������ƽ�����½���������Ϊ�ڻ�����֬��Ũ�Ƚ�С����Ӧ���Ż�����ײ��Ӧ�ļ��ʽ�С�����跴Ӧʱ��ϳ�������2.5 h�������ܲ�����ߣ�˵���䷴Ӧ�̶��Ѳ�����ߡ�
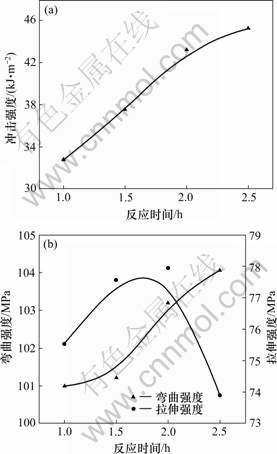
(a) ���ǿ�ȣ�(b) ����ǿ�Ⱥ�����ǿ��
ͼ5��DHBP��TDI��Ӧʱ��Թ��������ѧ���ܵ�Ӱ��
Fig.5 Effects of reaction time of DHBP and TDI on mechanical properties of mixture
2.3 �����;۰���/������֬������ϵ������ܷ���
2.3.1 ��ͬ����DHBP�Բ������ȶ��Ե�Ӱ��
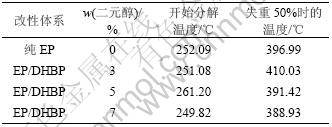
���ϵ��ȷֽ⡢ij�¶���ʧ���ʵȾ�Ϊ�����������ȶ��Ե���Ҫ��־��ͼ6��ʾΪDHBP���ԵĻ�����֬������ϵ�TG���ߣ���1��ʾΪ���ȷֽ��¶ȡ���ͼ6�ͱ�1��֪������DHBP���ԵĻ�����֬��TG�����봿������֬��TG�������غϣ���DHBP�����ϵ�ʱ���ȷֽ��¶���DHBP�������Ӷ����ߣ���DHBP������5%ʱ����ʼ�ȷֽ��¶�����Լ9 �棬����5%ʱ����Ȼ��ʼ�ֽ��¶������½������½����Ⱥ�С���ɼ�������DHBP������ʹ����ԵĻ�����֬��������н�ǿ�������ԡ�

w(DHBP): 1��0��2��3%��3��5%��4��7%
ͼ6��E51/DHBP������ϵ�TG����
Fig.6 TG curves of E51/DHBP blends
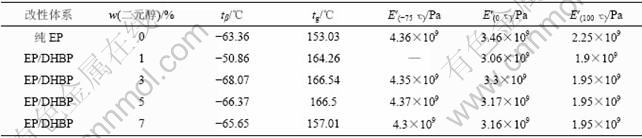
��1 �۰������Ի�����֬������ϵ��ȷֽ��¶�
Table 1 Decomposing temperature of polyurethane modified epoxy resin composites
2.3.2 DHBP�����Թ�����ϵIJ������¶ȵ�Ӱ��
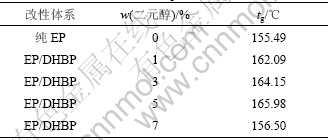
ͼ7��ʾΪDHBP���ԵĻ�����֬������ϵ�DSC���ߣ���2��ʾΪ�䲣�����¶ȡ���ͼ7�ͱ�2��֪��������3%~5% DHBPʱ��������֬��ϵ�IJ������¶�������ߣ��ȴ�������֬����Լ9 �档��Ȼ��TG��DSC��2�ַ����ķ��������һ�µģ�Ҳ��ǰ�����5%~6%�Ľ龧��������ʱ�����ϵ�����ǿ�ȡ�����ǿ����ͬʱ��ߵĽ����һ�¡������������ú��龧��Ԫ�ķ����;۰������Ի�����֬����һ����ȷ�Χ��������Ե�ͬʱ�������������֬����ѧ���ܺ������ܣ����Һ��н龧��Ԫ�ķ����;۰���/������֬������ϲ������¶���߷��Ƚϴ���ԭ���Ǿ��иհ�״�ṹ�Ľ龧�۰����������нϸߵ��۵�Ͳ�����ת���¶ȣ������н�ǿ�������ԡ�

w(DHBP): 1��0��2��1%��3��3%��4��5%��5��7%
ͼ7��E51/DHBP������ϵ�DSCͼ
Fig.7 DSC curves of E51/DHBP blends
��2�����龧��Ԫ�����;۰������Ի�����֬������ϵIJ������¶�tg
Table 2 Glass transition temperature of aromatic polyurethane containing mesomorphic unit modified epoxy resin composites
2.4 �����;۰���/������֬������϶�̬��ѧ����
2.4.1 DHBP�����Ի�����֬��������ɳ���ת���¶ȵ�Ӱ��
���̻���ϵ���ɳ��¶Ⱥ�ģ�����3��ʾ��ͼ8��ʾΪDHBP�����Ի�����֬����ģ����tan �ĵ�Ӱ�졣��ͼ8���Կ�������Ӧ�ڲ�����ת���ת���ֻ��1������Ϊ���壬��˵���������Ϊ������ϵ��DHBP�������ṹ����˫��A������֬�е�˫��A�ṹ���ƣ��Ҿ����뻷����֬���Ƶ��Ѽ����Σ����ɵķ����;۰����뻷����֬�����Ժã�����ͺ���ʱ�������뻷����֬�γ����Ӹ��ϡ�����3%~5% DHBPʱ����̬��ѧ���ԵĻ�����֬�IJ������¶����13.5 �档ͼ8(b)�Ц��ɳ���������ϵ���ǻ��ѻ��ţ����ϵ�������֬�����еĽ龧�۰������Ѽ����κͻ�����֬��ϵ�������ǻ��ѻ��Ź�ͬ���ף��ڵͺ�����Χ�ڣ�����DHBP���Ի�����֬�Ըı价����֬�н����������ǻ��ѵĺ������ʦ�ת���¶���������ƶ������Ƶ��仯���̻��Ĺ��������0 ����������ķ�(�¡� ת���)�������ڰ��̻���ϵ�����еġ�C��O��C�� ���������һ���µ��ɳ��˶�[3]��
��3 EP/DHBP������ϵ��ɳ��¶Ⱥ�ģ��
Table 3 Loosen temperature and modulus of EP/DHBP mixture

1��0%; 2��1%; 3��3%; 4��5%; 5��7%
ͼ8��EP/DHBP������ϵ�����ģ����tan ��
Fig.8 Storage modulus and tan �� of EP/DHBP mixture
2.4.2 DHBP�����Թ����������ģ����Ӱ��
��ͼ8�ͱ�3���Կ�������������ڵ����µĶ�̬����ģ���仯��С�����¶�����ʱ����ϵ������������ͣ������˶�������ǿ��������������������ӣ��������¶ȵ����ߣ���ϵ��ģ�����½����¶ȼ�������ʱ����ϵ�Ĵ���ģ����1����խ�ķ�Χ�ڽ���1�����ϵ�����������Ӧ���Dz���̬���ߵ�̬��ת�䡣
�ӱ�3���ɿ�������100 �����£�����Ԫ�������ϵ�ʱ������DHBP������֬��ģ�������½����½����Ⱥ�С������DHBP�����ļ������ӣ�������֬ģ���½����ȱ��Խ��ԽС����100 ��ʱ��ģ���������䡣ģ�������½���������ΪDHBP��Ȼ���н龧���ţ���DHBP�������ṹ�����Ѽ���֬�������������Ѽ�����ת���������ף�����һ������˳�ԣ���DHBPΪ�龧��Ԫ�ľ۰������Ի�����֬��ģ�������½��������Ⱥ�С����ˣ�������Ϊ�����龧��Ԫ�����;۰������Ի�����֬��������ϵ��ģ����
DHBP�������ӣ���Ӧ�IJ���̬��ߵ�̬ת�䣬������С��5%ʱ�������¶�����·����ƶ���DDM�̻��Ļ�����֬������С��̨�ף����밷�̻��Ĵμ�ת���йأ�����ϵ�й��е� ��C��O��C�� ���������һ���µ��ɳ��˶�[3]��
3 �� ��
a. ��DHBPΪԭ�ϣ�����ԭλ�ۺϵķ������ɺ��龧��Ԫ�ķ����;۰�������ʹ����̬�ľ۰������ȷ�ɢ�ڻ�����֬�У������Ӽ��ĸ����������ã����ƺ��龧��Ԫ�ķ����;۰��������������������֬�����Ե�ͬʱ���������֬����ѧģ�������龧����DHIP�ĺ���Ϊ6%��DHIP��TDIĦ����Ϊ1?2���̻���DDM����Ϊ35%����Ӧʱ��Ϊ2.5 hʱ��������ϵij��ǿ�����187.9%������ǿ�����4.7%������ǿ�����19.5%��
b. ������֬�������ֻ��1��������ת��壬Ϊ������ϵ����100 �����£��ڵͺ�����Χ�ڣ�����龧����������֬��ģ�������½����½����Ⱥ�С����龧���������ļ������ӣ����½����ȱ��Խ��ԽС����100��ʱ��ģ���������䡣��ת���¶ȱ仯����
c. ���龧��Ԫ�����;۰���/������֬��ϵΪ������ϵ����������Ҳ������ߣ�������5% DHBPʱ����ʼ�ȷֽ��¶ȺͲ������¶Ⱦ�����Լ9 �档ͻ���˴�ͳ���������Խ�����ǿ����������Ϊ���۵ľ� ���ԡ�
�ο����ף�
[1] �� ��. ���绷����֬��������״�뷢չ����[J]. ���ʻ�����Ϣ, 2002(1): 11-14.
HU Bin. Production status and developing trend of the worldwide epoxy resin[J]. Global Chemical Information, 2002(1): 11-14.
[2] ������, ֣�ļ�, �ܵ���. 2-��ϩ������-2-����������Ի�����֬��ˮ�Ի�[J]. ���ϴ�ѧѧ��: ��Ȼ��ѧ��, 2006, 37(1): 68-72.
MA Cheng-yin, ZHENG Wen-ji, ZHOU Di-wu. Waterborne epoxy resin modified by 2-acrylamido-2-methyle- propanesulfonic acid[J]. J Cent South Univ: Sci Technol, 2006, 37(1): 68-72.
[3] Ҧ����, �ɹ���. ������֬�����о���չ[J]. �ȹ�����֬, 2001, 16(2): 22-25.
YAO Kang-de, CHENG Guo-xiang. Research development of toughening epoxy resin[J]. Thermosetting Resin, 2001, 16(2): 22-25.
[4] ������, �� ��. Һ����Ȼ�����(CTBN)���ͻ�����֬���о�[J]. ���Ϲ���, 1995(5): 17-19.
ZHANG Yan-zhong, SHEN Chao. Study of toughened epoxy resin by using CTBN[J]. Journal of Materials Engineering, 1995(5): 17-19.
[5] �� ��, ������. ������֬�ĵ�����������[J]. ���ϿƼ�, 1999(5): 21-23.
WANG Xin, XUAN Zhao-long. Toughening modification of epoxy resins by elastomer[J]. Plastics Science and Technology, 1999(5): 21-23.
[6] ������, �� ��. �����Թ������϶Ի�����֬�������о�[J]. �ȹ�����֬, 1997, 12(4): 37-41.
DAI Li-zong, FU Xuan. Toughening modification of epoxy resin by engineering thermoplastics[J]. Thermosetting Resin, 1997, 12(4): 37-41.
[7] Bennett G S, Farris R J, Thompson S A. Amine-terminated poly(aryl ether ketone)-epoxy/amine resin systems as tough high performance materials[J]. Polymer, 1991, 32(9): 1633-1641.
[8] ���ı�, ��ϼ�. Һ��������֬������ͨ������֬���о�[J]. ճ��, 2000, 21(1): 17-20.
ZHONG Wen-bin, WANG Xia-yu. Study on properties of epoxy resin modified with liquid crystal epoxy resin[J]. Technology on Adhesion & Sealing, 2000, 21(1): 17-20.
[9] ����, �� ��, �����, ��. ������֬�����ϲ��ϵ��Ʊ�������[J]. �߷��Ӳ��Ͽ�ѧ�빤��, 2003, 19(2): 29-33.
HU You-hua, GAO Hui, QI Chen-ze, et al. A review of synthesis and properties of epoxy nanocomposites[J]. Polymer Materials Science & Engineering, 2003, 19(2): 29-33.
[10] ֣��Ƽ, ���ٲ�, ������. ������֬�������ϲ����о���չ[J]. �������Ͳ���, 2000(3): 17-18.
ZHENG Ya-ping, NING Rong-chang, QIAO Sheng-ru. Progress of epoxy-nanocomposites[J]. New Chemical Materials, 2000(3): 17-18.
[11] ������. ��������ۺ���(lPN)������չ����Ӧ��[J]. �����Ƽ�, 1989, 7(1): 28-34.
ZHANG Yong-ling. Development and Use about interpenetrating networks polymer[J]. Science and Technology of Lanzhou Chemistry Company, 1989, 7(1): 28-34.
[12] �� ��, ·̫ƽ. ������֬/�۰�������������о�[J]. �ȹ�����֬, 1996, 11(1): 13-16.
YU Hao, LU Tai-ping. Study on interpenetrating networks of epoxy resin/ polyurethane[J]. Thermosetting Resin, 1996, 11(1): 13-16.
[13] ��־��, ������, ������, ��. ԭλ�ۺ�PAM����EP���о�[J]. �߷��Ӳ��Ͽ�ѧ�빤��, 2000, 16(5): 71-74.
LIU Zhi-zhong, WANG Xin-lin, LOU Yin-pei, et al. Study on modification of epoxy resin through in-situ polymerization of PAM[J]. Polymer Materials Science & Engineering, 2000, 16(5): 71-74.
[14] �� Ӱ, ��С��. ԭλ�ۺϸ��Ը߷��Ӹ��Ի�����֬���о�[J]. �߷��Ӳ��Ͽ�ѧ�빤��, 1998, 14(5): 13-16.
ZHANG Ying, TANG Xiao-zhen. Study on modifying epoxy resin with rigid rod polymer formed by in-situ polymerization[J]. Polymer Materials Science & Engineering, 1998, 14(5): 13-16.
[15] Τ ��, ̷��ͥ, ������. ������Һ���ۺ�������Ի�����֬���ܵ�Ӱ���о�[J]. �й�����, 2001, 15(10): 34�C37.
WEI Chun, TAN Song-ting, LIU Min-na. Effect of kinds of liquid crystalline polymers on properties of epoxy resin[J]. China Plastics, 2001, 15(10): 34�C37.
[16] ������, �� ӱ. ������֬���������ϲ��ϵĿ���[J]. ճ��, 2001, 22(2): 4�C6.
WANG Hong-zuo, WANG Ying. Developments of epoxy resin-inorganic nanocomposites[J]. Technology on Adhesion & Sealing, 2001, 22(2): 4�C6.
[17] ������, ������. �۰���/������֬IPN���о�[J]. �������Ͳ���, 2001, 29(8): 21�C24.
HU Xiao-lan, LIANG Guo-zheng. Research of polyurethane and epoxy resin IPN[J]. New Chemical Materials, 2001, 29(8): 21�C24.
[18] Ф Ӿ. �ۺ���/ճ�������ϲ������½�չ [J]. ��������Ӧ��, 1998(8): 28�C30.
XIAO Yong. The latest advance of polymer/clay nanocomposities[J]. Application of Engineering Plastics, 1998(8): 28�C30.
[19] ���Ӷ�, �����, �� ��, ��. ��ճ������[M]. ����: ��ѧ��ҵ������, 2005, 2(1): 18.
LI Zi-dong, LI Guang-yu, JI Li, et al. Adhesives additives[M]. Beijing: Chemical Industry Press, 2005.
�ո����ڣ�2007-10-08�������ڣ�2007-12-11
������Ŀ������ʡ�ص�Ƽ��ƻ�������Ŀ(2006GK2022)
ͨ�����ߣ�¬ ��(1964-)���У����ϳ����ˣ���ʿ�����»�����֬�̻������о��������绰��0731-8836319��E-mail: luoffice@mail.csu.edu.cn

