


Trans. Nonferrous Met. Soc. China 22(2012) 286-390
Structural, magnetic and electronic properties of FeF2 by first-principle calculation
YANG Zhen-hua1, 2, 3, WANG Xian-you1, 2, 3, LIU Li1, 2, 3, SU Xu-ping3
1. Faculty of Materials, Optoelectronics and Physics, Xiangtan University, Xiangtan 411105, China;
2. School of Chemistry, Xiangtan University, Xiangtan 411105, China;
3. Key Laboratory of Materials Design and Preparation Technology of Hunan Province, Xiangtan University, Xiangtan 411105, China
Received 8 April 2011; accepted 16 June 2011
Abstract: First-principle calculation was used to investigate the magnetic properties, electronic structure and bonding mechanism of FeF2. By calculating the lattice parameters and magnetic moment as a function of effective interaction parameter (Ueff), it is found that the optimum value of Ueff is equal to 4 eV, the magnetic moment is 3.752 ��B and the value of c/a is 0.704, which are in good agreement with the experiment results. Simultaneously, on the basis of GGA+U method, the electronic structure and bonding mechanism of FeF2 were investigated by the analysis of electron localization function, Bader charge and total charge density. The results show that the bonding behavior between Fe and F atoms is a combination of ionic and covalent bond.
Key words: magnetic moment; electronic structure; bonding mechanism; FeF2; lithium-ion battery
1 Introduction
Lithium-ion batteries as power source have been widely used in various energy storage fields due to high energy density and good cycling stability [1, 2]. The commonly used cathode materials for lithium-ion batteries are the layered intercalation compounds, such as LiCoO2, LiMnO2, LiFePO4.
Recently, metal fluorides are considered next generation lithium ion battery cathode materials and enter a prominent space in energy storage [3-5]. The reaction between Li and metal fluorides can be expressed by the following equation:
 (1)
(1)
where Me is metal, such as Fe, Co, Ni, Bi; e is the charge of an electron. Due to the high ion bonding character between metal and high electro-negativity of F ions and full use of their valence electrons in charge/discharge process, the chemical energy can be reversibly utilized in metal fluorides through conversion reaction (1), which results in high average voltage and high specific capacity. For example, the theoretical average voltages of CrF3 and FeF2 are 2.28 and 2.66 V, respectively [6, 7].
Among all metal fluorides, FeF2 with theoretical voltage of 2.66 V is considered a promising cathode material for the application of lithium ion battery owing to its low cost and low toxicity. In order to analyze the intrinsic electronic structure of FeF2, theoretical calculations on FeF2 have also been carried out. BROWN et al [8] found that the Fe-F interactions presented some covalent bond characters by means of DV-X�� method. The magnetic coupling constants between nearest-neighbor magnetic ions in FeF2 were calculated using Hartree-Fock method [9]. PHILIPP et al [10] calculated the energy gap and spin-magnetic moments for FeF2 by using the generalized gradient approximation (GGA) and local-spin-density approximation (LSDA) methods. The result shows that LSDA calculation fails (making them metallic) and GGA calculation yields improvement on the energy gap over LSDA calculation, but the energy gap is very small (0.4 eV), which cannot describe the wide band gap character state of FeF2. Besides, the spin-magnetic moment calculated by GGA calculations (3.5 ��B) is lower than the experimental result (3.75 ��B).
Based on the above discussions, because on-site coulomb interaction exists between Fe 3d and GGA method is not enough to well describe the wide band gap character of the compound, the introduction of a Hubbard-like contribution on the Fe 3d states is necessary. Hence, GGA+U method is put forward. U is the Hubbard parameter, which reflects the strength of the on-site coulomb interaction. Up to date, no attempt was carried out on using GGA+U method to study the structure of FeF2. In this work, first-principle calculation within GGA+U method was used to investigate the magnetic and electronic properties as well as bonding mechanism of FeF2.
2 Computational method
The calculations were performed using the ab-initio total-energy and molecular-dynamics program Vienna ab-initio simulation program (VASP) developed at the institut f��r Materialphysik of the Universit?t Wien [11-14]. The projector augmented wave (PAW) method was implemented in VASP. The approximation used for exchange and correlation energies is GGA. For GGA calculations, the Perdew-Burke-Ernzonhof (PBE) exchange-correlation functional was chosen. To study the strong correlation character of 3d electrons of Fe, GGA+U method was employed. Within the GGA+U approach, the on-site coulomb term U and the exchange term J were considered together. Effective interaction parameter (Ueff) can be concluded by U-J. In order to obtain the appreciate value of U, the electronic structure of FeF2 was investigated with the U parameters changing from 1 to 10 eV. The U value was determined through the comparison of equilibrium cell parameters with the available experimental data. The J value was fixed as 1 eV. The cutoff energy of 520 eV and Monkhorst-Pack grid (6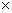 69) for k point sampling in the first irreducible Brillouin zone were used to ensure the good convergence for total energy and forces acting on the atoms. All atoms were fully relaxed until the Hellman-Feynman forces on the atoms were converged to less than 1��10-3 eV/nm by a Quasi Newton (QN) algorithm method.
69) for k point sampling in the first irreducible Brillouin zone were used to ensure the good convergence for total energy and forces acting on the atoms. All atoms were fully relaxed until the Hellman-Feynman forces on the atoms were converged to less than 1��10-3 eV/nm by a Quasi Newton (QN) algorithm method.
To obtain the electronic density of states (DOS), the tetrahedron method with Bl?chl corrections was used for the Brillouin-zone integration and a 8��8��12 Monkhorst-Pack k-point mesh was set. In all calculations, spin-polarized calculations were performed.
3 Results and discussion
3.1 Crystal structure of FeF2
FeF2 has a tetragonal structure (with two formula units per unit cell) with space group P42/mnm. Usually, it can be described by 3 structural parameters: a, c and internal parameter u. a and c are 0.46945 and 0.33097 nm, respectively. The positions of the atoms inside the unit cell are (0, 0, 0) and (1/2, 1/2, 1/2) for the Fe atoms, and (u, u, 0), (1-u, 1-u, 0), (1/2+u, 1/2-u, 1/2) and (1/2-u, 1/2+u, 1/2) for the F atoms, where u is 0.301 [15]. After the structure of FeF2 is fully relaxed, u is 0.299.
3.2 Ferromagnetic and antiferromagnetic phase of FeF2
The dependence of the total energies on Ueff for ferromagnetic (FM) and antiferromagnetic (AFM) phase is calculated and shown in Fig. 1. As seen from Fig. 1, with the increase of U values, the energy difference between AFM and FM phases decreases; the energy difference between AFM and FM phases is negligible when Ueff value is higher than 3.0 eV. Generally speaking, the total energy of AFM phase is lower than that of FM phase, which indicates that AFM phase is more stable than FM phase. Hence, in the following calculations, only the AFM phase is considered in investigating the magnetic and electronic properties of FeF2.
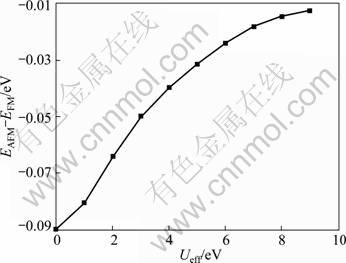
Fig. 1 Energy difference between AFM phase and FM phase (EAFM-EFM) within GGA+U as function of Ueff
3.3 Geometry and magnetic properties
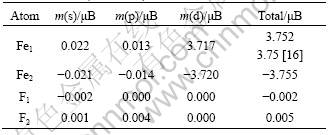
The calculated lattice constant a and experimental value are compared in Fig. 2. As seen from Fig. 2, it is clear that the GGA+U method overestimates the lattice constant, and the error increases with the increase of Ueff value. Even though the theoretical error is below 1.5% compared with experimental value, the calculated lattice constant is still in reasonable agreement with the experiment. Figure 3 shows the tendency of c/a value as a function of Ueff. It can be found that the value of c/a decreases with the increase of Ueff value, and it is the closest to the experimental value at Ueff = 4 eV. Moreover, a series of Ueff values are employed to calculate the magnetic moment of Fe2+ in FeF2. The calculated results are shown in Fig. 4. It is observed from Fig. 4 that the magnetic moment of Fe2+ increases with the increase of Ueff, and it is 3.752 ��B at Ueff=4 eV, which is in good agreement with the reported experimental value of 3.750 ��B [16]. In order to further understand the magnetic properties of FeF2, the calculated spin density is plotted for the FeF2  plane using GGA+U (Ueff=4 eV) method, as shown in Fig. 5. According to spin-up and spin-down electrons, Fe atoms are classified as Fe1 and Fe2, and F atoms are classified as F1 and F2, respectively. As seen from Fig. 5, it is obvious that the magnetic properties of FeF2 are mainly contributed from the magnetic moment of Fe atoms. As for F atoms, they exhibit very small induced spin magnetic moment by Fe atoms. The calculated spin magnetic moments of Fe and F atoms of FeF2 are listed in Table 1. Apparently, the spin magnetic moment of Fe is chiefly contributed by Fe 3d state. Moreover, it is interesting that a small spin polarization on the site of F atoms appears, indicating that Fe1-Fe2 superexchange interaction occurs in virtue of F atoms.
plane using GGA+U (Ueff=4 eV) method, as shown in Fig. 5. According to spin-up and spin-down electrons, Fe atoms are classified as Fe1 and Fe2, and F atoms are classified as F1 and F2, respectively. As seen from Fig. 5, it is obvious that the magnetic properties of FeF2 are mainly contributed from the magnetic moment of Fe atoms. As for F atoms, they exhibit very small induced spin magnetic moment by Fe atoms. The calculated spin magnetic moments of Fe and F atoms of FeF2 are listed in Table 1. Apparently, the spin magnetic moment of Fe is chiefly contributed by Fe 3d state. Moreover, it is interesting that a small spin polarization on the site of F atoms appears, indicating that Fe1-Fe2 superexchange interaction occurs in virtue of F atoms.
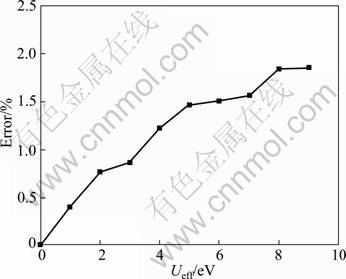
Fig. 2 Errors between calculated lattice constant and experimental value

Fig. 3 Ratio of c and a parameters (c/a) within GGA+U as function of Ueff
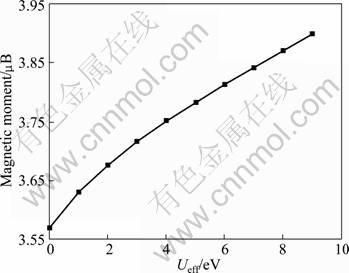
Fig. 4 Magnetic moment within GGA+U as function of Ueff
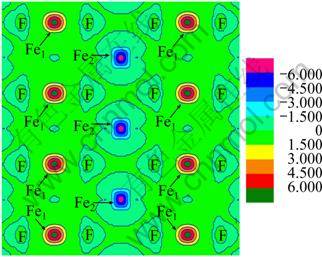
Fig. 5 Spin density of FeF2 AFM phase in  plane in GGA+U (Ueff =4)
plane in GGA+U (Ueff =4)
Table 1 Atomic orbit magnetic moments in FeF2 crystal cell within GGA+U (Ueff =4 eV)
3.4 Electronic structure of FeF2
The calculated total DOS together with the partial density of states (PDOS) of FeF2 using the GGA and GGA+U(Ueff =4) methods are compared in Fig.6. It can be seen that the band gap calculated by GGA method is close to 0. The inclusion of correlation effect Ueff=4.0 eV for FeF2 can push the states at Fermi level away from Fermi level and reproduce the wide band gap character of the compound. The GGA+U calculations show that FeF2 belongs to Mott-Hubbard materials with a band gap of 2.565 eV. Therefore, the GGA+U calculations can better predict the electronic structure of FeF2 than GGA calculation. Further analysis on the DOS of FeF2 is carried out. The valence band is divided into two regions. One is located between -7.34 and -1.05 eV, which shows significant hybridization of Fe3d and F2p. The other is located between -1.05 eV and Fermi level, which is composed of mainly Fe3d and small F2p. Besides, the conduction band is located between 2.57 and 8.28 eV.
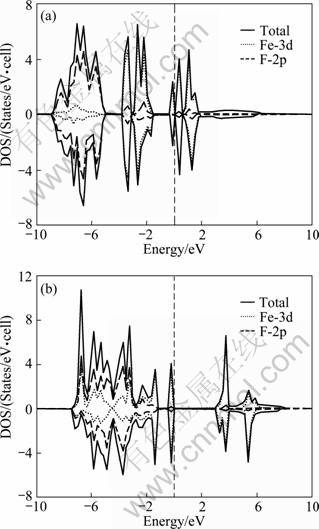
Fig. 6 Total and partial DOS for FeF2 AFM phase within GGA (a) and GGA+U (b) (Ueff=4 eV) (Fermi level was set 0, positive and negative DOS represents spin up and down DOS, respectively.)
In order to analyze the bonding mechanism of FeF2, the charge density distribution of FeF2 is plotted in Fig. 7. It is found that the charge density around Fe and F ions is spherical distributed with slightly directional character toward the direction to their nearest neighboring atoms, indicating that a little covalent bond exists between Fe and F atoms.
The bonding mechanism can be also further analyzed by electron localization function (ELF) [17, 18]. The value of ELF ranges from 0 to 1.0 with ELF=0.5 corresponding to the free electron gas distribution. An ELF value equal to 1.0 means perfect electron localization. The calculated ELF maps in the plane of FeF2 are given in Fig.8. The ELF values of interstitial region between Fe and F atoms are small but none zero, which indicates the bonding between Fe and F atoms processes ionic nature. Hence, the bonding behavior between Fe and F atoms is the combination of covalent bonding and ionic interactions.
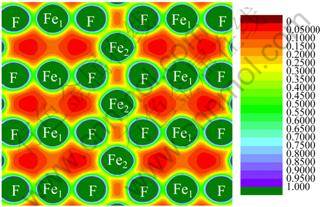
Fig. 7 Calculated total charge density of FeF2 AFM phase in plane within GGA+U at Ueff =4 eV
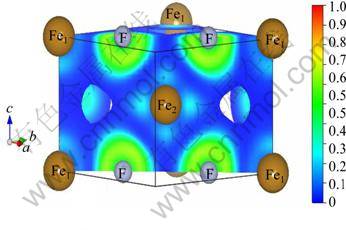
Fig. 8 Calculated plots of ELF of FeF2 AFM phase within GGA+U at Ueff =4 eV
To describe the covalent and ionic nature quantitatively, the net charges on Fe and F ions are calculated using Bader��s AIM theory [19, 20]. The results are listed in Table 2. As seen from Table 2, it is evident that about 1.5503 electrons transfer from each Fe atom to F atoms. However, net charge on Fe and F atoms are less than nominal charges of the ions (+2 for Fe and -1 for F). Accordingly, in FeF2, the bond between Fe and F atom shows evident ionic character and a little covalent bond exists between Fe and F atoms at the same time.
Table 2 Net charge on atoms of Fe and F in FeF2 calculated using Bader charge analysis

4 Conclusions
1) FeF2 AFM phase is more stable than FeF2 FM phase considering the strongly correlated Fe 3d, which is consistent with the experimental results. It is well known that GGA calculations result in a metallic state, while GGA+U method can well describe the wide band gap character of FeF2 and the calculated band gap is 2.565 eV.
2) The optimum value of Ueff is equal to 4 eV, the magnetic moment is 3.752 ��B, and the value of c/a is 0.704, which is in good agreement with the experimental results.
3) It can be concluded that the bond between Fe and F atoms in FeF2 is a mixture of ionic bond and covalent bond.
References
[1] DO Jing-shan, DAI Rui-feng. Cobalt oxide thin ?lm prepared by an electrochemical route for Li-ion battery [J]. J Power Sources, 2009, 189(1): 204-210.
[2] YAN Feng, LI Ya-li, HOU Feng. Preparation and electrochemical properties of Cr doped LiV3O8cathode for lithium ion batteries [J].Mate Lett, 2009, 63(15):1338-1340.
[3] YAMAKAWA N, JIANG M, KEY B, Grey C P. Identifying the local structures formed during lithiation of the conversion material, iron fluoride, in a Li ion battery: A solid-state NMR, X-ray diffraction, and pair distribution function analysis study [J]. J Am Chem Soc, 2009, 131(30): 10525-10536.
[4] LIAO P, McDONALD B L, DUNLAP R A, DAHN J R. Combinatorially prepared [LiF]1-xFex nanocomposites for positive electrode materials in Li-ion batteries [J]. Chem Mater, 2008, 20(2): 454-461.
[5] LI T, LI L, CAO Y L, Ai X P, YANG H X. Reversible three-electron redox behaviors of FeF3 nanocrystals as high-capacity cathode-active materials for Li-ion batteries [J]. J Phys Chem C, 2010, 114(7): 3190-3195.
[6] LI H, BALAYA P, MAIER J. Li-storage via heterogeneous reaction in selected binary metal fluorides and oxides [J]. Electrochem Soc A, 2004, 151(11): 1878-1885.
[7] AMATUCCI G G, PEREIRA N. Fluoride based electrode materials for advanced energy storage devices [J]. J Fluorine Chem, 2007, 128(4): 243-262.
[8] BROWN P J, FIGGIS B N, REYNOLDS P A. Covalency in FeF2 from magnetisation density data and from ab initio theory [J]. J Phys: Condens Mat, 1990, 2(24): 5297-5308.
[9] MOREIRA I D P R, DOVESI R, ROETTI C, SAUNDERS V R, ORLANDO R. Ab initio study of MF2 (M=Mn, Fe, Co, Ni) rutile-type compounds using the periodic unrestricted Hartree-Fock approach [J]. Phys Rev B, 2000, 62(12): 7816-7823.
[10] PHILIPP D, PETER B, VLADIMIR S, KARLHEINZ S. Generalized-gradient-approximation description of band splittings in transition-metal oxides and fluorides [J]. Phys Rev B, 1994, 49(15): 10170-10175.
[11] KRESSE G, HAFNER J. Ab initio molecular-dynamics simulation of the liquid-metal�Camorphous-semiconductor transition in germanium [J]. Phys Rev B, 1994, 49(20): 14251-14269.
[12] KRESSE G, FURETHM?LLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Comput Mater Sci, 1996, 6(1): 15-50.
[13] KRESSE G, FURETHM?LLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Phys Rev B, 1996, 54(16): 11169-11186.
[14] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method [J]. Phys Rev B, 1999, 59(3): 1758-1775.
[15] BAUR W H, KHAN A A. Rutile-type compounds SiO2, GeO2 and a comparison with other rutile-type structures [J]. Acta Crystallographica B, 1971, 27(1): 2133-2139.
[16] DOE R E, PERSSON K A, MENG Y S, CEDER G. First-principles investigation of the Li-Fe-F phase diagram and equilibrium and nonequilibrium conversion reactions of iron fluorides with lithium [J]. Chem Mater, 2008, 20(16): 5274-5283.
[17] MATAR S F, DEMAZEAU G J. Ab initio studies of the electronic structure of the quaternary system LiBC4N4 [J]. J Alloys Compd, 2009, 182(1-2): 2678-2679.
[18] OSORIO-GUILL?N J M, SIMAK S I, WANG Y, JOHANSSON B, AHUJA R. Bonding and elastic properties of superconducting MgB2 [J]. Solid State Commun, 2002, 123(6-7): 257-262.
[19] HENKELMAN G, ARNOLDSSON A, J?NSSON H. A fast and robust algorithm for Bader decomposition of charge density [J]. Comput Mater Sci, 2006, 36(3): 354-360.
[20] DELLE SITE L. A statistical interpretation of Bader��s definition of interatomic surface [J]. Phys Lett A, 2001, 286(1): 61-64.
��һ��ԭ������FeF2�Ĵ��ԡ��ṹ����������
����1, 2, 3, ������1, 2, 3, �� ��1, 2, 3, ����ƽ3
1. ��̶��ѧ ������������ѧԺ����̶ 411105��
2. ��̶��ѧ ��ѧѧԺ����̶ 411105��
3. ��̶��ѧ ������Ƽ��Ʊ���������ʡ�ص�ʵ���ң���̶ 411105
ժ Ҫ�����õ�һ��ԭ�������о�FeF2�Ĵ��ԡ����ӽṹ�ͳɼ�������ͨ�����㲻ͬUֵ�µĵľ�������ʹžأ������ѵ���ЧUֵ(Ueff)Ϊ4 eV����ʱ����Fe�Ĵžغ;������c��a �ı�ֵ�ֱ�3.752 ��B��0.704����ʵ�����Ǻϡ����⣬����GGA+U���������öԵ��Ӿ�������Bader��ɺ��ܵ�ɵķ������о�FeF2 �ĵ��ӽṹ�ͳɼ��������������Fe��F֮�京�����Ӽ����ۼ���������
�ؼ��ʣ��žأ����ӽṹ���ɼ�������FeF2������ӵ��
(Edited by FANG Jing-hua)
Foundation item: Project (20871101) supported by the National Natural Science Foundation of China; Project (09C945) supported by the Scientific Research Fund of Hunan Provincial Education Department, China
Corresponding author: WANG Xian-you; Tel: +86-731-58292060; Fax: +86-731-58292061; E-mail: wxianyou@yahoo.com
DOI: 10.1016/S1003-6326(11)61188-6

