
?Adsorption of atomic S and C on Mg(0001) surface
HU Yu-lin(������)1, ZHANG Wei-bing(������)1, TANG Bi-yu(�Ʊ���)1,
DING Wen-jiang(���Ľ�)2, ZENG Xiao-qin(��С��)2
1. Key Laboratory of Advanced Materials and Rheological Properties of Ministry of Education,
Department of Physics, Xiangtan University, Xiangtan 411105, China;
2. State Key Laboratory of Metal Matrix Composite,
School of Materials Science and Engineering, Shanghai Jiaotong University, Shanghai 200030��China
Received 10 April 2006; accepted 25 April 2006
Abstract: First-principle calculations based on density functional theory were used to study the adsorption of atomic sulfur and carbon on the Mg(0001) surface in a wide range of coverages from 1/4 ML(monolayer) to 1 ML. It is found that the adsorption of atomic S and C on the high coordinate hollow site is more energetically favorable than that on other adsorption sites. S atom is favorable to be adsorbed at on-surface site and C atom is favorable to be adsorbed at subsurface site. The results suggest that when the coverage increases, the binding energy for S and C atoms will decrease and the interaction between adsorbed atoms tends to be stronger. It indicates that as coverage increases, S-Mg and C-Mg interaction weakens.
Keywords: adsorption; chemisorptions; density function theory; generalized gradient approximation; high coordinate; charge density
1 Introduction
In modern industry, Mg and Mg alloys are widely used for their excellent properties of low specific gravity, high specific strength and high recycle rate. Sulfur and carbon are the common impurities present in magnesium and magnesium alloys; they affect the bulk and surface properties of the metal. Millions of dollars are lost every year due to the negative effects of sulfur and carbon poisonings.
It is often difficult in experimental studies to control the type and level of sulfur and carbon contaminants, making it hard to determine how a specific adsorption process is taken place. Using a theoretical approach, the process of sulfur and carbon atoms adsorption on low-index surface can be determined precisely. Recently, a number of theoretical studies on adsorption of sulfur [1-9] and carbon [10-13] on a series of metal surfaces have been published, but to our knowledge, no first-principle calculation for atomic sulfur and carbon adsorption at magnesium surface has been performed so far.
In this work, S and C atoms adsorbed on magnesium surface were investigated. The main goal is to study the similarities and differences between sulfur and carbon bonding on the magnesium substrates, and the most favored configurations formed by the adsorbed sulfur and carbon atoms at three different coverages.
2 Computational details
This study is based on the density functional theory using the generalized gradient approximation (GGA) for the exchange-correlation functional. The Vienna ab initio Simulation Package (VASP) [14-16] is used in the calculation. For the GGA exchange-correlation function, the PW91 [17, 18] scheme was used which gives satisfactory results in many studies of adsorptions on metal surfaces. The core-electron interactions were described by ultrasoft-pseudopotentials [19] for Mg, S and C, and expand one-electron Kohn-Sham [20] states in a plane wave basis with kinetic energies up to a cutoff of 300 eV. For all surfaces we considered, super cell geometry with periodic boundary conditions was used, and all calculations were performed nonspin-polarized.
As a test of the accuracy of our approach, the calculated lattice constants for hcp Mg is a=0.319 nm and c/a=1.63, which agree well with the experimental values (a=0.321 nm and c/a=1.623) [21] and other GGA computational data [22,23], the bulk modulus, ignoring lattice vibrations, is found to be 35.7 GPa, in good agreement with the measured values 35.5GPa [24] and other computational data (34.1 GPa) [23]. Our calculated bond lengths are 0.190 0 nm and 0.127 5 nm for S2 and C2, respectively, which are reasonably good comparing with corresponding 0.192 nm [25] and 0.124 3 nm [26] from experiments.
The clean Mg(0001) surface was modeled by periodic slabs consisting of seven magnesium layers separated by ten layers thickness(about 2.6 nm) of vacuum, with a Monkhorst-Pack k-point mesh of 16��16��1 for (1��1) surface, 8��8��1 for (2��2) surface and 8��16��1 for 2��1 surface. This slab thickness, vacuum thickness, and the chosen energy cutoff were shown to give well-converged binding-energy values, the error of total energy was less than 1 meV/atom, so it can give a precise description for this system. The S atoms are adsorbed on one side of the slab; a Fermi-surface smearing of 0.2 eV is applied to the Brillouin zone integrations. The positions of Mg atoms in the three topmost magnesium layers and all of the adsorbed S or C atoms are fully optimized until the sum of the Hellmann-Feynman forces on all unconstrained atoms converges to less than 0.2 eV/nm. For the clean Mg metal surface the relaxations of the interlayer spacing with respect to the bulk spacing we calculated show 1.6% expansion, 0.25% contraction and 0.9% contraction of the first, second, and third interlayer distance respectively, agree well with the experiment data and other computational data [22]. The calculated work function is 3.72 eV for the relaxed surface also agrees well with the experimental value 3.84 eV and the calculated value (3.72 eV) [22].
In the present calculations, all the binding energies reported in this study were calculated with respect to the unspin-polarized free atomic S and C. More formally, the binding energy (Ebind) for different sites was calculated by:
Ebind=(EMg(0001)+N��ES(C)-ES(C)/Mg(0001) )/N (1)
where ES(C)/Mg(0001) represents the total energy of the sulfur or carbon atom covered Mg(0001) slab, EMg(0001) is the total energy of a clean, relaxed Mg(0001) slab, and ES(C) is the total energy of an isolated sulfur or carbon atom, N represents the number of absorbed sulfur or carbon atoms.
3 Results and discussion
As shown in Fig.1, four high-symmetry binding sites (top, bridge, hcp, and fcc) on Mg(0001) surface were considered. Table 1 summarizes the binding energies calculated for the S and C atoms in the more
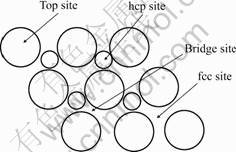
Fig.1 Schematic top view of Mg (0001) surface showing position of adsorption sites of top, bridge, hcp and fcc site. The large circles represent top layer Mg atoms and the small circles represent the second layer Mg atoms
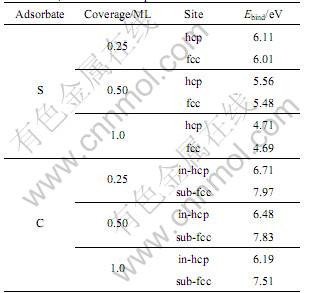
Table 1 Binding energy(Ebind) for adsorption of S atoms in hcp and fcc sites, C atoms in in-hcp and sub-fcc sites
energetically favored site for different coverages.
The calculations reveal that high coordinate site hcp and fcc are the favorable site for both S and C atoms. Furthermore, it can be found that the binding energy for both S and C atoms decreases when coverage increases, but there has an intrinsic difference. For sulfur atoms, on-surface hcp site is the most favored site followed by on-surface fcc site which is slightly less stable, for C atoms the fcc site about halfway between the surface and the second layer (sub-fcc) is the most favored site followed by the hcp site slightly below the surface (in-hcp), where in-hcp site is the ideal tetrahedral hollow site and sub-fcc is the ideal octahedral hollow site. When the carbon atom is initiated at on-surface site at lower coverages(����0.5 ML), it will sink into the subsurface during energy minimization.
The earlier researches for sulfur adsorbed on close-packed transition metal surfaces indicate that on hcp metals the stable adsorption site is hcp and on fcc metals the favorable site is fcc [8]. The present results also manifest this feature.
For on-surface hcp site, when sulfur coverage increased from 0.25 ML to 0.50 ML and 1 ML, the binding energy decreased significantly from 6.11 eV to 5.56 eV and 4.71 eV respectively, the binding energy for fcc site also decreased from 6.01 eV to 5.48 eV and 4.69 eV respectively, it can be found that at 1.00 ML coverage, the binding energy difference between hcp and fcc site is only 0.02 eV. In order to make a comparison, in-hcp and sub-fcc sites adsorption of sulfur atoms also have been calculated, the results reveal that the binding energy is much smaller than on-surface site.
For carbon atoms, at sub-fcc site, binding energy decreased from 7.97 to 7.51 eV when coverage increased from 0.25 ML to 1 ML, at in-hcp site the binding energy decreased from 6.71 to 6.19 eV. When coverage increases to 1 ML, the binding energy of on-surface fcc and hcp sites also have been calculated, it is found that at on-surface sites the binding energies are 2.5-1.5 eV less than that of sub-fcc and in-hcp sites.
The shortest bond distance is an important parameter that can be used to compare adsorption character for different sites. This parameters is indicated by labels d(S�DMg) and d(C�DMg), the results are shown in Fig.2.
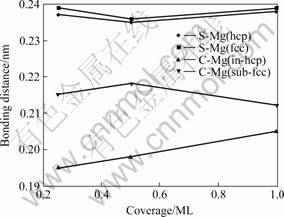
Fig.2 Shortest bonding distance in different coverages for S atom in hcp and hcp site, C atom in in-hcp and sub-fcc site
For S adsorption, the shortest S�DMg bond distance was determined to be 0.237, 0.235, 0.238 for hcp site at 0.25 ML, 0.50 ML and 1.00 ML coverage respectively, and for fcc site the bonding distance is about 0.004-0.001 nm longer, related with the phenomena of the binding energy at fcc site is slightly less than hcp site. For comparison, the bonding distance for top site also were calculated, it is found that the bonding distance is about 0.010-0.003 nm shorter than hcp and fcc site. At the top site S atom is directly bonding to only one atom and at the hollow sites the bonding is distributed over more atoms. It can be found that these distances are again related to the coordination number of the adsorption site.
For C atom adsorptions, at sub-fcc site, bonding distance is 0.215, 0.218 and 0.212 nm for 0.25 ML, 0.5 ML and 1 ML coverage respectively. For in-hcp site the bonding distance is 0.195, 0.198 and 0.204 nm respectively. Whereas in-hcp is a fourfold-coordinated tetrahedral site and sub-fcc is a sixfold-coordinated octahedral site, the present result also proves that C atom preferred to stay at high-coordinate site.
After adsorption of S or C atoms, the calculations suggest that in stable sites both relaxation and reconstruction occurs as the unconstrained Mg atoms moved in the x-, y-, and z-directions. For S adsorption, wherever S atoms are adsorbed, the distance of Mg atom movement in x- and y-direction is less than 1.5% compared to the Mg lattice constant, and for C adsorption, the Mg atom movement in x- and y-direction is less than 4% compared to the Mg lattice constant.
Another interesting observation is that at some coverages and sites, the different degrees of atomic relaxation within the same layer cause a buckling of the layer. The bucklings of the first Mg layer compared to the Mg layer thickness in various coverages are listed in Table 2.
Table 2 Buckling of the first Mg layer(Buck1) after adsorption compared to Mg layer thickness
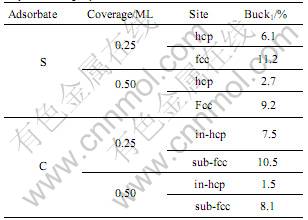
In general, the magnitude of the buckling of the first Mg layer (Buck1) decreases from the first layer to second layer and third layer as the effect of the adsorbate is weaken, and there has no buckling when coverage reaches 1 ML. From Table 2, it can be found that at lower coverages (����0.5 ML), the buckling for S atom adsorption at fcc site is more bigger than at hcp site; for C adsorption, the C atom induces more bigger buckling when it is adsorbed at sub-fcc site.
Calculations of the change in work function (����) of the Mg(0001) surface after adsorption can give an indication of the charge reorganization which affects the surface dipole moment. In general, a decrease in the work function after adsorption indicates a transfer of charge from the adsorbate to the substrate, while an increase in the work function indicates a transfer of charge from the substrate to the adsorbate.
The calculated work function changes after S or C atoms adsorption in different sites and coverages were determined by subtracting the calculated work function value of the clean relaxed surface from those of the S or C adsorbed surface values. From calculation, it is found that for on-surface S adsorption, there has a sharply increase in work function, at hcp site, when S coverage increases from 0.25 ML to 0.50 ML and 1 ML, the change of work function ���� is 0.87, 1.57 and 2.30 eV respectively, and for fcc site the change is 0.91, 1.66 and 2.30 eV, respectively, indicating that the S is always behaving as an electropositive species. For carbon atoms adsorbed at in-surface site and subsurface site, there has a very slightly negative changes in work function for all coverages, both of in-hcp and sub-fcc site, the changes of work function are in range of -0.02--0.11 eV.
4 Conclusions
1) The adsorptions of atomic S and C on Mg(0001) were investigated using the first-principle DFT-GGA calculations. Three coverages (0.25 ML, 0.5 ML and 1.0 ML) were considered.
2) For S adsorption, on-surface hcp site is the most favorable adsorption site, followed by fcc site with a little difference. For C adsorption, the results show that subsurface site is much more energetically favored than on-surface site, and the sub-fcc site is the most favored site. The on-surface adsorption of S atoms shows sharply decrease of binding energy and continuous increase of work function with the increase of coverage, indicating that the S is behaving as an electronegative species.
3) For C adsorption, at sub-fcc and in-hcp site, when the coverage increases, the binding energy also decreases but not so much compared with S adsorption, and the work function decreases slightly for all considered coverages.
References
[1] Alfonso D R. First-principles study of sulfur overlayers on Pd(111) surface [J]. Sur Sci, 2005, 596(1-3): 229-241.
[2] Sokolowski M, Pfn��r H. Continuous order-disorder phase transitions of the p(2��2) and (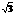 �� )R30? superstructures of sulfur on Ru(001): Effective critical exponents and finite-size effects [J]. Phys Rev B, 1994, 49(11): 7716-7728.
�� )R30? superstructures of sulfur on Ru(001): Effective critical exponents and finite-size effects [J]. Phys Rev B, 1994, 49(11): 7716-7728.
[3] Spencer M J S, Snook I K, Yarovsky I. Coverage-dependent adsorption of atomic sulfur on Fe(110): A DFT study [J]. J Phys Chem B, 2005, 109(19): 9604-9612.
[4] Nelson S G, Spencer M J S, Snook I K, Yarovsky I. Effect of S contamination on properties of Fe(100) surfaces [J]. Sur Sci, 2005, 590(1): 63-75.
[5] L��decke J, Ettema A R H F, Driver S M, Scragg G, Kerkar M, Woodruff D P, Cowie B C C, Jones R G, Bastow S. The structure of sulphur adsorption phases on Ni(111) studied by X-ray standing wavefield absorption [J]. Sur Sci, 1996, 366(2): 260-274.
[6] Rodriguez J A, Dvorak J, Jirsak T, Liu G, Hrbek J, Aray Y, Gonz��lez C. Coverage effects and the nature of the metal-sulfur bond in S/Au(111): High-resolution photoemission and density-functional studies [J]. JACS, 2003, 125(1): 276-285.
[7] Yang Z x, Wu R q, Rodriguez J A. First-principles study of the adsorption of sulfur on Pt.111: S core-level shifts and the nature of the Pt-S bond [J]. Phys Rev B, 2002, 65(15): 155409- 01-155409-09.
[8] Lahtinen J, Kantola P, Jaatinen S, Habermehl-Cwirzen K, Salo P, Vuorinen J, indroos M, Pussi K, Seitsonen A P. LEED and DFT investigation on the (2��2)-S overlayer on Co(0001) [J]. Sur Sci, 2005, 599(1-3): 113-121.
[9] Rodriguez J A, Chaturvedi S, Kuhn M. The adsorption of sulfur on Rh(111) and Cu/Rh(11) surfaces [J]. J Chem Phys, 1998, 108(7): 3064-3073.
[10] Furthm��ller J, Kresse G, Hafner J, Stumpf R, Scheffler M. Site-Selective adsorption of C atoms on Al(111) surface [J]. Phys Rev Lett, 1995, 74(25): 5084-5087.
[11] Zhang Q M, Wells J C, Gong X G, Zhang Z y. Adsorption of a carbon atom on the Ni38 magic cluster and three low-index nickel surfaces: A comparative first-principles study [J]. Phys Rev B, 2004, 69(20): 205413-01-205413-7.
[12] Jiang D E, Carter E A. Carbon atom adsorption on and diffusion into Fe(110) and Fe(100) from first principles [J]. Phys Rev B, 2005, 71(4): 045402-1-045402-6.
[13] Neyman K M, Inntam C, Gordienko A B, Yudanov I V, R?sch N. Adsorption of carbon on Pd clusters of nanometer size: A first-principles theoretical study [J]. J Chem Phys, 2005, 122(17): 174705-1-174705-9.
[14] Kresse G, Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Phys Rev B, 1996, 54(16): 11169-11186.
[15] Kresse G, Furthmuller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Comput Mater Sci, 1996, 6(1): 15-50.
[16] Kresse G, Hafner, J. Ab initio molecular dynamics for open-shell transition metals [J]. Phys Rev B, 1993, 48(17): 13115-13118.
[17] Perdew J P, Wang Y. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation [J]. Phys Rev B, 1986, 33(12): 8800-8802.
[18] Perdew J P, Chevary J A, Vosko S H, Jackson K A, Pederson M, Singh D J, Fiolhais C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation [J]. Phys Rev B, 1992, 46(11): 6671-6687.
[19] Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Phys Rev B, 1990, 41(11): 7892-7895.
[20] Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects [J]. Phys Rev, 1965, 140(4A): 1133-2238.
[21] Hardie D, Parkins R N. Lattice spacing relationships in magnesium solid solutions [J]. Philos Mag, 1959, 4(43): 815-825.
[22] Schr?der E, Fasel R, Kiejna A. O adsorption and incipient oxidation of the Mg(0001) surface [J]. Phys Rev B, 2004, 69(11): 115431 -1-115431-8.
[23] Wachowicz E, Kiejna A. Bulk and surface properties of hexagonal-close-packed Be and Mg [J]. J Phys Condens Matter, 2001, 13(48): 10767-10776.
[24] Wazzan A R, Robinson L B. Elastic constants of magnesium-lithium alloys [J]. Phys Rev, 1967, 155(3): 586-594.
[25] Huber K P, Herzberg G. Molecular Spectra and Molecular Structure IV: Constants of Diatomic Molecules Van Nostrand-Reinhold [M]. New York, 1974.
[26] Radzig A A, Smirnov B M. Reference Data on Atoms, Molecules, and Ions [M]. Berlin: Springer-Verlag, 1985.
(Edited by YANG Hua)
Corresponding author: TANG Bi-yu; Tel: +86-732-2371004; Fax: +86-732-8292468; E-mail: tangbiyu@xtu.edu.cn

