Mg-Al-Ca-Sn合金中二元相金属间化合物结构稳定性、电子结构、弹性性质和热力学性质的第一性原理计算
来源期刊:中国有色金属学报(英文版)2016年第1期
论文作者:王峰 孙士杰 于波 张峰 毛萍莉 刘正
文章页码:203 - 212
关键词:Mg-Al系合金;第一性原理计算;电子结构;弹性性质;热力学性质
Key words:Mg-Al alloy; first-principles calculation; electronic structure; elastic properties; thermodynamic properties
摘 要:通过第一性原理计算方法研究Mg-Al-Ca-Sn合金中主要强化相Mg17Al12、Al2Ca、Mg2Sn和Mg2Ca的结构稳定性、电子结构、弹性常数和热力学性质。计算所得晶格常数与实验值及文献值吻合。合金形成热和结合能计算结果表明,Al2Ca具有最强的合金形成能力和结构稳定性。通过对这些化合物的态密度、Mulliken电子占据数、金属性和差分电荷密度计算分析其结构稳定性机制。通过计算 Mg17Al12、Al2Ca、Mg2Sn和Mg2Ca 的弹性常数,推导出各相的体模量、剪切模量、弹性模量和泊松比。热力学性质计算结果表明,Al2Ca和Mg2Sn的Gibbs自由能低于Mg17Al12,即Al2Ca和Mg2Sn 的晶体结构稳定性优于Mg17Al12相。因此,通过添加Ca和Sn元素可以提高Mg-Al系合金的热力学稳定性。
Abstract: The structural stability, electronic structures, elastic properties and thermodynamic properties of the main binary phases Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca in Mg-Al-Ca-Sn alloy were determined from the first-principles calculation. The calculated lattice parameters are in good agreement with the experimental and literature values. The calculated heats of formation and cohesive energies show that Al2Cahas the strongest alloying ability and structural stability. The densities of states (DOS), Mulliken electron occupation number, metallicity and charge density difference of these compounds are given. The elastic constants of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca phases are calculated, and the bulk moduli, shear moduli, elastic moduli and Poisson ratio are derived. The calculations of thermodynamic properties show that the Gibbs free energies of Al2Ca and Mg2Sn are lower than that of Mg17Al12, which indicates that Al2Ca and Mg2Sn are more stable than Mg17Al12 phase. Hence, the heat resistance of Mg-Al-based alloys can be improved by adding Ca and Sn additions.
Feng WANG, Shi-jie SUN, Bo YU, Feng ZHANG, Ping-li MAO, Zheng LIU
School of Materials Science and Engineering, Shenyang University of Technology, Shenyang 110870, China
Received 29 March 2015; accepted 2 July 2015
Abstract: The structural stability, electronic structures, elastic properties and thermodynamic properties of the main binary phases Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca in Mg-Al-Ca-Sn alloy were determined from the first-principles calculation. The calculated lattice parameters are in good agreement with the experimental and literature values. The calculated heats of formation and cohesive energies show that Al2Ca has the strongest alloying ability and structural stability. The densities of states (DOS), Mulliken electron occupation number, metallicity and charge density difference of these compounds are given. The elastic constants of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca phases are calculated, and the bulk moduli, shear moduli, elastic moduli and Poisson ratio are derived. The calculations of thermodynamic properties show that the Gibbs free energies of Al2Ca and Mg2Sn are lower than that of Mg17Al12, which indicates that Al2Ca and Mg2Sn are more stable than Mg17Al12 phase. Hence, the heat resistance of Mg-Al-based alloys can be improved by adding Ca and Sn additions.
Key words: Mg-Al alloy; first-principles calculation; electronic structure; elastic properties; thermodynamic properties
1 Introduction
As the lightest metallic structural material, magnesium alloys are widely used in automotive, aerospace, electronic equipment and other fields [1-3]. Especially, Mg-Al alloy system is the most widely used magnesium alloy because aluminum, as the most favorable alloying element, improves casting fluidity, hot-crack resistance and mechanical properties of magnesium without significantly raising its density. However, the poor elevated strength is a major obstacle to its wider structural applications because of the Mg17Al12 phase at grain boundary, which softens and coarsens at temperatures exceeding 120 °C [4,5]. Hence, a lot of effort has been devoted to improve the mechanical properties of Mg-Al-based alloys. It has been shown that the mechanical properties of magnesium alloys could be improved remarkably by the alloying effect [6,7]. Due to the low cost, calcium and tin have been used to improve the poor heat resistance property of Mg-Al-based alloys, and there has been increasing attention to the development of the Mg-Al-Ca-Sn- based alloys [8]. To the best of our knowledge, calcium was added to the Mg-Al-based alloys, in which both creep and damping capacity were significantly improved [9,10]. LIN et al [11] had studied the improvement of the mechanical properties at elevated temperature of die-cast AZ91 alloy with Ca addition by forming Al2Ca. The heat of formation, cohesive properties and density of states of Al2Ca and Mg2Ca were also calculated in their research. However, the hot-crack resistance of AZ91D alloy decreased with Ca addition [12]. Fortunately, the hot-crack resistance of AZ91 magnesium alloy increased with Sn addition [13]. Hence, Sn element could be chosen to improve the hot-crack resistance of Mg-Al-Ca alloys. Furthermore, RASHNO et al [14] and MAHMUDI et al [15] had studied the microstructure and creep behavior of magnesium alloys with Ca and Sn additions. They reported that the improvement on creep resistance for these alloys was mainly attributed to the formation of thermally stable intermetallics, which provided the pinning effect at the magnesium grain boundaries. YU et al [16] had calculated the electronic structure and elastic properties of AB2 type Laves phase in Mg-Al-Ca alloy to study the underlying mechanism of structural stability and mechanical properties. ZHONG et al [17] had studied the first-principles calculation of three Laves phase structures in Al2Ca and Al2Mg compositions to predict the existence of Al2(Mg,Ca) Laves phase. MAO et al [18] had studied the structure stability, electronic structure and elastic property of the intermetallics of Sn alloying Mg-Zn-Ca alloy. However, no systematic theoretical study has been reported on electronic structures, elastic properties and thermodynamics of binary intermetallics in Mg-Al-Ca- Sn alloys.
In this work, we have carried out the first-principles theoretical calculations to investigate the electronic structures, elastic properties and thermodynamic properties of the binary intermetallics in Mg-Al-Ca-Sn alloy. The results give a valuable estimation for the properties unavailable in experiments, which provide theoretical basis for the development of a new type of magnesium alloy.
2 Computational methods
Cambridge sequential total energy package (CASTEP), a first-principles plane wave pseudo- potentials method based on density function theory (DFT), was used for the calculations [19]. Generalized gradient approximation (GGA) of the Perdew-Burke- Ernzerhof (PBE) was used to describe the exchange- correlation energy function [20,21]. The ultrasoft pseudo-potential was used to describe the interaction between ion-core and valence electron [22]. The parameters affecting the calculation accuracy are kinetic energy cutoff and the number of k-points network in Brillouin zone, and the cut-off energy of plane wave was set as 380 eV, the k-points separations for Mg17Al12, Al2Ca and Mg2Sn are 6×6×6 and that for Mg2Ca is 6×6×4. The conjugate-gradient algorithm was used for lattice structure optimization. Relaxation and self- consistent calculations of lattice constants and atomic positions was carried out by using the Broyden- Fletcher-Goldfarb-Shanno (BFGS) method. Geometry optimization was carried out under the electron relaxation until the total energy convergence was 5.0×10-8 eV/atom, and the force on all atoms was less than 0.01 eV/A. The stress-strain method was used to calculate the elastic constants of Mg17Al12, Al2Ca, Mg2Ca and Mg2Sn. The maximum strain amplitude was set as 0.003. The total energy convergence was 1.0×10-6 eV/atom, the maximum force was 0.002 eV/A, and the maximum displacement is 1.0×10-4 A.
3 Results and discussion
3.1 Crystal structure and lattice constant
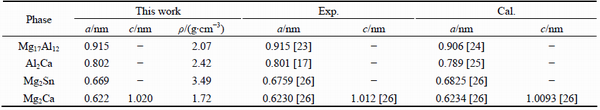
The structure parameters and lattice constants of these phases are listed in Tables 1 and 2. The calculated lattice constants are in good agreement with the experimental and other theoretical values, and the error is only 2.0%. Hence, the calculated results in this work are highly reliable.
Table 1 Structure parameters and atomic coordinates of binary phases in Mg-Al-Ca-Sn alloys

Table 2 Equilibrium crystal parameters (a, c) and density (ρ) of binary phases in Mg-Al-Ca-Sn alloys
3.2 Heats of formation and cohesive energies
Based on the related theories of the first-principle calculation, a lower formation enthalpy means stronger alloying ability of a phase, and a lower cohesive energy indicates a more stable crystal lattice [27,28]. The formation enthalpy ( ) is defined to decompose the crystal into bulk pure elements and expressed by the following equation:
) is defined to decompose the crystal into bulk pure elements and expressed by the following equation:
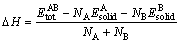 (1)
(1)
where 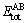 is the total energy of compound representing phases A and B, NA and NB are the atom numbers in the unit cell,
is the total energy of compound representing phases A and B, NA and NB are the atom numbers in the unit cell,  and
and 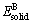 are the energies per atom of pure elements A and B, respectively. The calculated energies of Mg, Al, Ca and Sn in solid states are -973.9493, -56.4195, -1001.4756 and -95.4475 eV/atom, respectively. The calculated heats of formation of the binary phases in Mg-Al-Ca-Sn alloys are listed in Table 3.
are the energies per atom of pure elements A and B, respectively. The calculated energies of Mg, Al, Ca and Sn in solid states are -973.9493, -56.4195, -1001.4756 and -95.4475 eV/atom, respectively. The calculated heats of formation of the binary phases in Mg-Al-Ca-Sn alloys are listed in Table 3.
Table 3 Heat of formation ( (Ecoh) of binary phases in Mg-Al-Ca-Sn alloys

The calculated heats of formation are in good agreement with experimental results and other theoretical values, which confirms reliability of our calculation method. Further analysis found that heats of formation of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca are all negative, which confirms that the structure of these phases exists stably. Furthermore, it can be concluded that Al2Ca phase has the strongest alloying ability, next Mg2Sn, Mg2Ca and finally Mg17Al12.
Whereas, cohesive energy Ecoh is defined to decompose the crystal into single free atoms and expressed by the following equation:
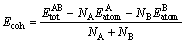 (2)
(2)
where  and
and refer to the energy per atom under freedom state. The calculated energies of Mg, Al, Ca and Sn in free states are -972.4847, -52.7251, -999.5694 and -91.0698 eV/atom, respectively. The obtained results are also listed in Table 3.
refer to the energy per atom under freedom state. The calculated energies of Mg, Al, Ca and Sn in free states are -972.4847, -52.7251, -999.5694 and -91.0698 eV/atom, respectively. The obtained results are also listed in Table 3.
Further analysis found that Al2Ca phase has the largest cohesive energy, and far more than those of Mg2Sn, Mg17Al12 and Mg2Ca. In addition, Mg2Sn phase has higher cohesive energy than Mg17Al12, which indicates that Mg2Sn phase is more stable than Mg17Al12. All the analysis results indicate that adding Ca or Sn to Mg-Al alloys can promote the structural stability by forming Al2Ca and Mg2Sn phases, which agrees well with the reported experimental results [14,15].
3.3 Electronic structures
In the present work, the electronic structures are calculated to have a further understand of the bonding of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca, and then to reveal the potential structural stability mechanism of these phases. The total and partial densities of states (DOS) of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca are presented in Fig. 1. From Fig. 1(a), it can be found that the main bonding peaks of Mg17Al12 between -50 and -40 eV mainly originate from the contribution of valence electron number of Mg(p) and Al(p) orbits, while the main bonding peaks between -10 and 5 eV are the contribution of Al(s), Al(p), Mg(s) and Mg(p). From Fig. 1(b), the main bonding peaks of Al2Ca are located at three energy ranges. The main bonding peaks between -45 and -40 eV are dominated by the valence electron number of Ca(s), while the main bonding peaks between -25 and -20 eV are the contribution of Ca(p) and Al(p) orbits. In addition, the main bonding peaks between -10 and 10 eV are the contribution of Ca(s), Ca(p), Ca(d), Al(s) and Al(p). From Fig. 1(c), the main bonding peaks of Mg2Sn between -45 and 40 eV mainly originate from the contribution of valence electron number of Mg(p) orbits, while the contributions of valence electron between -10 and 15 eV are Mg(s), Mg(p), Sn(s) and Sn(p). From Fig. 1(d), the main bonding peaks of Mg2Ca are located in three energy ranges. The main bonding peaks between -45 and -40 eV are dominated by the valence electron numbers of Ca(s) and Mg(p), while the main bonding peaks between -25 and -20 eV mainly originate from the contribution of valence electron number of Ca(p) orbits. In addition, the main bonding peaks between -10 and 5 eV are the contribution of Ca(s), Ca(d), Mg(s) and Mg(p). Further comparison of total densities of states (DOS) of these compounds per atom near the Fermi level is performed as shown in

Fig. 1 Total and partial DOS of Mg17Al12 (a), Al2Ca (b), Mg2Sn (c) and Mg2Ca (d) and total DOS of binary phases in Mg-Al-Ca-Sn alloys per atom (e)
The Mulliken electron occupation numbers of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca are listed in Table 4. It is found that the charges of Mg transfer to Al in Mg17Al12, the transfer number (per atom) is 0.513; for Al2Ca, the charges transfer from Ca atoms to Al atoms, the transfer number (per atom) is 0.92; for Mg2Sn, the charges transfer from Mg atoms to Sn atoms, the transfer number (per atom) is 0.50; for Mg2Ca, the charges transfer from Ca atoms to Mg atoms, the transfer number (per atom) is 0.90. The results indicate that the ionicity of these compounds increases in the following sequence: Mg2Sn< Mg17Al12
Table 4 Mulliken electron occupation numbers of Mg 17Al12, Al2Ca, Mg2Sn and Mg2Ca

The metallicity of the compound is estimated by the following equation [30]:
 (3)
(3)
where DF is the DOS value at the Fermi level; T is the temperature; kB is the Boltzmann constant; nm and ne are the thermal excited electrons and valence electron density of the cell, respectively; ne is calculated by ne=N/Vcell; N is the total number of valence electrons and Vcell is the cell volume. The related parameters and calculated results are shown in Table 5. It can be observed that fm increases in the following sequence: Mg2Sn< Al2Ca< Mg2Ca< Mg17Al12.
Table 5 Density of states at Fermi level (DF), total number of valence electrons (N), cell volume (Vcell) and metallicity parameter (fm) of binary phases in Mg-Al-Ca-Sn alloys

In order to further reveal the covalent and ionic bonding characteristics, the charge density difference was investigated. Charge density difference, which is defined as the electron density difference between the isolated atoms and their bonding states, can directly reflect the bonding characteristics. The results are shown in Fig. 2. The contour lines are plotted from -0.35 to 0.35 e/A3 with 0.15 e/A3 interval. In Fig. 2(a), the bonding between Al and its adjacent Al atom is mainly covalent, the bonding between Al and Mg is ionic and the bonding between Mg and Mg is metallic. In Fig. 2(b), the bonding between Al and the nearest Al atom is covalent, the bonding between Al and Ca is ionic and the bonding between Ca and Ca is metallic. In Fig. 2(c), it is found that there are covalent Sn―Sn bonds, ionic Mg―Sn bonds and metallic Mg―Mg bonds. In Fig. 2(d), metallic Mg―Mg bonds, ionic Mg―Ca bonds and covalent Ca―Ca bonds are found in Mg2Ca. Furthermore, the bonding characteristics of Mg17Al12 and Mg2Ca are found to be dominantly ionic, and the bonding characteristics of Al2Ca and Mg2Sn are found to be dominantly covalent. Based on the above discussion, it can be found that Al2Ca and Mg2Sn have stronger structural stability than Mg17Al12, which agrees well with the above calculated results of the heat of formation and the cohesive energy.
3.4 Elastic properties
Elastic constants relate to the deformation resistant capacity to an externally applied stress. Cubic crystal has three independent elastic constants namely C11, C12 and C44, the corresponding stability criteria [31] are: C11>0, C11>|C12|, C44>0, C11+2C12>0. Hexagonal crystal has six elastic constants (C11, C12, C13, C33, C44 and C66), and only five of them are independent since C66= (C11-C12)/2, the corresponding stability criteria [32] are: C11>0, C11-C12>0, C44>0, C66>0 and (C11+C12)C33-2 >0.The elastic properties of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca, which are important for manufacturing Mg-Al-based alloys with Ca and Sn addition, will be briefly discussed in this part. The calculated elastic constants for these phases in the present work are tabulated in Table 6.
>0.The elastic properties of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca, which are important for manufacturing Mg-Al-based alloys with Ca and Sn addition, will be briefly discussed in this part. The calculated elastic constants for these phases in the present work are tabulated in Table 6.
Mg17Al12, Al2Ca and Mg2Sn phases belong to cubic crystals. The bulk modulus (B) and shear modulus (G) of Mg17Al12, Al2Ca and Mg2Sn phases were calculated as follows [37]:
 (4)
(4)
 (5)
(5)
Mg2Ca phase belongs to hexagonal crystals. The bulk modulus (B) and shear modulus (G) of Mg2Ca phases were calculated as follows [37]:
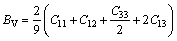 (6)
(6)
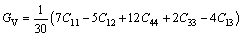 (7)
(7)
 (8)
(8)

Fig. 2 Electron density difference of Mg17Al12 (a), Al2Ca (b), Mg2Sn (c) and Mg2Ca (d)
Table 6 Calculated elastic constants of binary phases in Mg-Al-Ca-Sn alloys

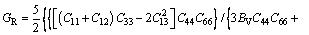
 (9)
(9)
 (10)
(10)
 (11)
(11)
The Poisson ratio (ν) and elastic modulus (E) of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca phases were deduced according to the following equations [37]:
 (12)
(12)
 (13)
(13)
The calculated mechanical parameters of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca are listed in Table 7. The bulk modulus (B) of a substance measured the substance’s resistance to applied pressure [38]. The larger the bulk modulus is, the stronger the capacity of the resist deformation is. From Table 7, it can be concluded that the largest bulk modulus of Al2Ca has the strongest resistance to volume change by applied pressure, next Mg17Al12 and Mg2Sn, finally Mg2Ca. Similarly, the shear modulus is a measure of resist reversible deformation by shear stress [38]. The larger the resistance is, the stronger the capacity of the resist shear deformation is. The calculated results demonstrate that Al2Ca has the largest resistance, followed by Mg17Al12, Mg2Sn and Mg2Ca. Hence, the deformation resistant capacity of Al2Ca would be much stronger than that of Mg17Al12, Mg2Sn and Mg2Ca. In addition, Poisson ratio was used to quantify the stability of the crystal against shear, which usually ranges from -1 to 0.5. The bigger the Poisson ratio is, the better the plasticity is. From Table 7, the calculated results demonstrate that Mg2Sn has the best plasticity because of the largest Poisson ratio, next Mg2Ca and Mg17Al12, finally Al2Ca. Elastic modulus is defined as the ratio of stress to strain, and it can be used to provide a measure of the stiffness of the solid. The larger the E is, the stiffer the material is. The calculated results in Table 7 indicate that the stiffness of Al2Ca is the largest, then Mg17Al12 and Mg2Sn, finally Mg2Ca.
Table 7 Bulk modulus (B), shear modulus (G), elastic modulus (E), elastic constants (Cij), G/B, and Poisson ratio (ν) of binary phases in Mg-Al-Ca-Sn alloys

The ratio of shear modulus to bulk modulus (G/B) of polycrystalline phases was used to predict the brittle and ductile behavior of materials [38]. The critical value, which is used to separate brittleness from ductility, is about 0.57. The G/B values of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca are 0.625, 0.773, 0.551 and 0.565, respectively. So, Mg2Sn and Mg2Ca are ductile, Mg17Al12 and Al2Ca are brittle. Furthermore, the calculated G/B for Al2Ca is the largest, indicating that Al2Ca is the most brittle among the four phases. The ductility or brittleness of crystal is also determined by its C12-C44 value [39]. The phase with positive value is ductile, otherwise, it is brittle. Hence, ductile phases are Mg2Sn and Mg2Ca, while Mg17Al12 and Al2Ca are thought as brittle phases.
3.5 Thermodynamics stabilities
The thermodynamic properties of the binary phases in Mg-Al-Ca-Sn alloys are calculated in this section. As a fundamental parameter, the Debye temperature (ΘD) correlates with many physical properties of solids, such as elastic constants, melting temperature and specific heat. At low temperatures, the Debye temperature was calculated from elastic constants, which are got from the earlier calculation [40], since ΘD may be calculated from the average sound velocity (vm) via the following equation [41]:
 (14)
(14)
where  is the Planck’s constant; kB is the Boltzmann constant; nA is the Avogadro number; n is the total number of atoms per formula; ρ is the density; M is the relative molecular mass per formula. The average wave velocity in the polycrystalline material can be approximately calculated by using the following equation:
is the Planck’s constant; kB is the Boltzmann constant; nA is the Avogadro number; n is the total number of atoms per formula; ρ is the density; M is the relative molecular mass per formula. The average wave velocity in the polycrystalline material can be approximately calculated by using the following equation:
 (15)
(15)
where vl and vt are longitudinal and transverse sound velocities, respectively, which are gained from the Hill’s bulk modulus and shear modulus from the Navier’s equation [42]:
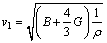 (16)
(16)
 (17)
(17)
The calculated results of ΘD, vl and vt are listed in Table 8. The calculated Debye temperature agrees well with the reported theoretical values. The largest ΘD is 498.88 K for Al2Ca. The ΘD is used to characterize the strength of covalent bonds in solids. From Table 8, it can be concluded that the covalent bond in Al2Ca is stronger than that of other phases. Hence, the mechanical and thermal stability of Al2Ca is the best one among four phases, which agrees with the earlier calculated elastic constant.
To study the structure stability of the binary phases in Mg-Al-Ca-Sn alloys at different temperatures, the global phonon spectrum is calculated, since the knowledge of the global phonon spectrum of a given
Table 8 Theoretically calculated thermal properties of binary phases in Mg-Al-Ca-Sn alloys including longitudinal sound velocity, shear sound velocity and average sound velocity, and Debye temperature

system enables the investigation of its significant thermodynamic quantities and the relative stability of its various phases at different temperatures [46]. The enthalpy E(T), entropy S(T) and free energy F(T) are given by applying the following formula [47]:
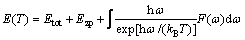 (18)
(18)

 (19)
(19)

 (20)
(20)
where Etot is the total energy, Ezp is the zero point energy, is the Planck’s constant, F(ω) is the phonon density of states and T is temperature. In this work, the free energy (F) is equal to the Gibbs free energy (G) because the calculations were carried out under 0 GPa. The enthalpy, entropy and Gibbs free energy for the binary phases in Mg-Al-Ca-Sn alloys as a function of temperature are shown in Fig. 3.
It can be seen from Fig. 3(a) that the enthalpy increases with increasing temperature. At temperatures up to 222 K, the enthalpies have the following sequence: Al2Ca>Mg2Sn>Mg2Ca>Mg17Al12. In the range of 222-283 K, the growth trend of enthalpies H is Al2Ca> Mg2Ca>Mg2Sn>Mg17Al12. And in the range of 283-1000 K, the growth trend of H is Al2Ca> Mg2Ca>Mg17Al12 > Mg2Sn. From Fig. 3(b), it can be seen that the entropy increases gradually with increasing temperature. When the temperature is lower than 60 K, the values of entropies (S) have the following sequence: Mg2Sn> Al2Ca>Mg2Ca>Mg17Al12. In the range of 60-1000 K, the values of S have the following sequence: Al2Ca>Mg2Sn> Mg2Ca>Mg17Al12. From Fig. 3(c), it is found that the Gibbs free energy gradually decreases with increasing temperature. It is noted that, when the temperature is lower than 100 K, the growth trend of G is: Mg17Al12>Mg2Ca>Al2Ca>Mg2Sn. In the range of 100-1000 K, the values of Gibbs free energy have the following sequence: Mg17Al12>Mg2Ca>Mg2Sn>Al2Ca. The smaller the Gibbs free energy is, the better the thermal stability of compounds is [29]. Hence, the calculated Gibbs free energies show that the thermal stability of these compounds gradually increases in the order of Mg17Al12, Mg2Ca, Mg2Sn and Al2Ca phases in the range of 273-573 K. The thermal stabilities of Al2Ca and Mg2Sn phases are better than that of Mg17Al12 phase and do not change with the elevated temperature, that is to say, Ca and Sn additions to the Mg-Al alloys can improve the heat resistance by forming Al2Ca and Mg2Sn phases, which can well provide the theoretical basis for the development of a new type of heat resistant magnesium alloy from the point of view of alloy energy.

Fig. 3 Thermodynamic properties of vibrational enthalpy (a), entropy (b) and Gibbs free energy (c) for binary phases in Mg-Al-Ca-Sn alloys
4 Conclusions
1) The calculated lattice parameters are in good agreement with the experimental and literature values. The calculated heats of formation and cohesive energies show that Al2Ca has the strongest alloying ability.
2) The calculation of bonding electron number shows that Al2Ca has the strongest structural stability, then Mg2Sn and Mg17Al12, finally Mg2Ca. The metallicity of the compound is estimated. Charge density difference can directly reflect the bonding characteristics. The bonding characteristics of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca are all covalent bonds, ionic bonds and metallic bonds.
3) The elastic constants of Mg17Al12, Al2Ca, Mg2Sn and Mg2Ca phases are calculated. The results of bulk modulus, shear modulus, elastic modulus and Poisson ratio show that Al2Ca has the strongest resist deformation capacity, the plasticity of Mg2Sn phase is the best, and the stiffness of Al2Ca is the largest. Mg2Sn and Mg2Ca are ductile, and Mg17Al12 and Al2Ca are brittle.
4) The calculated Debye temperature agrees well with the reported theoretical and experimental values. The calculated Gibbs free energy shows that the thermal stability of these compounds gradually increases in the order of Mg17Al12, Mg2Ca, Mg2Sn and Al2Ca.
References
[1] MORDIKE B L, EBERT T. Magnesium: Properties-applications- potential [J]. Materials Science and Engineering A, 2001, 302(1): 37-45.
[2] MUSTAFA K K. Magnesium and its alloys applications in automotive industry [J]. The International Journal of Advanced Manufacturing Technology, 2008, 39(9-10): 851-865.
[3] AGNEW S R, NIE J F. Preface to the viewpoint set on: The current state of magnesium alloy science and technology [J]. Scripta Materialia, 2010, 63(7): 671-673.
[4] LUO A A. Recent magnesium alloy development for elevated temperature applications [J]. International Materials Reviews, 2004, 49(1): 13-30.
[5] KONDORI B, MAHMUDI R. Impression creep characteristics of a cast Mg alloy [J]. Metallurgical and Materials Transactions A, 2009, 40(8): 2007-2015.
[6] ZHANG Y C, YANG L, DAI J, GUO G L, LIU Z. Effect of Ca and Sr on microstructure and compressive creep property of Mg-4Al-RE alloys [J]. Materials Science and Engineering A, 2014, 610: 309-314.
[7] KIM B H, LEE S W, PARK Y H, PARK I M. The microstructure, tensile properties, and creep behavior of AZ91, AS52 and TAS652 alloy [J]. Journal of Alloys and Compounds, 2010, 493(1-2): 502-506.
[8] KIM B H, LEE S W, PARK Y H, PARK I M. Microstructure, tensile properties and creep behavior of Mg-4Al-2Sn containing Ca alloy produced by different casting technologies [J]. Materials Science and Engineering A, 2012, 535: 40-47.
[9] KONDORI B, MAHMUDI R. Effect of Ca additions on the microstructure, thermal stability and mechanical properties of a cast AM60 magnesium alloy [J]. Materials Science and Engineering A, 2010, 527(7-8): 2014-2021.
[10] JUN J H. Damping behaviors of as-cast and solution-treated AZ91-Ca magnesium alloys [J]. Journal of Alloys and Compounds, 2014, 610: 169-172.
[11] LIN L, WANG F, YANG L, CHEN L J, LIU Z, WANG Y M. Microstructure investigation and first-principle analysis of die-cast AZ91 alloy with calcium addition [J]. Materials Science and Engineering A, 2011, 528(15): 5283-5288.
[12] TANG B, LI S S, WANG X S, ZENG D B, WU R. Effect of Ca/Sr composite addition into AZ91D alloy on hot-crack mechanism [J]. Scripta Materialia, 2005, 53(9): 1077-1082.
[13] TUREN Y. Effect of Sn addition on microstructure, mechanical and casting properties of AZ91 alloy [J]. Materials and Design, 2013, 49: 1009-1015.
[14] RASHNO S, NAMI B, MIRESMAEILI S M. Impression creep behavior of a cast MRI153 magnesium alloy [J]. Materials and Design, 2014, 60: 289-294.
[15] MAHMUDI R, MOEENDARBARI S. Effects of Sn additions on the microstructure and impression creep behavior of AZ91 magnesium alloy [J]. Materials Science and Engineering A, 2013, 566: 30-39.
[16] YU W Y, WANG N, XIAO X B, TANG B Y, PENG L M, DING W J. First-principles investigation of the binary AB2 type Laves phase in Mg-Al-Ca alloy: Electronic structure and elastic properties [J]. Solid State Sciences 2009, 11(8): 1400-1407.
[17] ZHONG Y, LIU J, WITT R A, SHON Y H, LIU Z K. Al2(Mg,Ca) phases in Mg-Al-Ca ternary system: First-principles prediction and experimental identification [J]. Scripta Materialia 2006, 55(6): 573-576.
[18] MAO P L, YU B, LIU Z, WANG F, JU Y. First-principles calculation of electronic structure and elastic property of AB2 type intermetallics in Mg-Zn-Ca alloy [J]. Acta Metallurgica Sinica, 2013, 49(10): 1227-1233.
[19] SHI D M, WEN B, MELNIK R, YAO S, LI T J. First-principles studies of Al-Ni intermetallic compounds [J]. Journal of Solid State Chemistry, 2009, 182(10): 2664-2669.
[20] PERDEW J P, BURKE K, EREZERHOF M. Generalized gradient approximation made simple [J]. Physical Review Letters, 1996, 7(18): 3865-3868.
[21] MARLO M, MILMAN V. Density-functional study of bulk and surface properties of titanium nitride using different exchange- correlation functionals [J]. Physical Review B, 2000, 62(4): 2899-2907.
[22] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Physical Review B, 1990, 41(11): 7892-7895.
[23] MIN X G, DU W W, XUE F, SUN Y S. The melting point of Mg17Al12 phase improved by alloyed with Ca and analysis based on the empirical electron theory (EET) [J]. Chinese Science Bulletin, 2002, 47(2): 109-112.
[24] ZHOU D W, LIU J S, XU S H, PENG P. First-principles investigation of the binary intermetallics in Mg-Al-Sr alloy: Stability, elastic properties and electronic structure [J]. Computational Materials Science, 2014, 86: 24-29.
[25] ZHOU D W, LIU J S, PENG P, CHEN L, HU Y J. A first-principles study on the structural stability of Al2Ca, Al4Ca and Mg2Ca phases [J]. Materials Letters, 2008, 62(2): 206-210.
[26] ZHANG H, SHANG S L, SAAL J E, SAENGDEEJING A, WANG Y, CHEN L Q, LIU Z K. Enthalpies of formation of magnesium compounds from first-principles calculations [J]. Intermetallics, 2009, 17(11): 878-885.
[27] MEDVEDEVA M I, GORNOSTYREV Y N, NOVIKOV D L, MRYASOV O N, FREEMAN A J. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl [J]. Acta Materialia, 1998, 46(10): 3433-3442.
[28] SAHU B R. Electronic structure and bonding of ultralight LiMg [J]. Materials Science and Engineering B, 1997, 49(1): 74-78.
[29] HONG S Y, FU C L. Phase stability and elastic moduli of Cr2Nb by first-principles calculations [J]. Intermetallics, 1999, 7(1): 5-9.
[30] LI Y F, GAO Y M, XIAO B, MIN T,FAN Z J,MA S Q,XU L L. Theoretical study on the stability, elasticity, hardness and electronic structures of W-C binary compounds [J]. Journal of Alloys and Compounds, 2010, 502(1): 28-37.
[31] YU W Y, WANG N, XIAO X B, TANG BY, PENG L M, DING W J. First-principles investigation of the binary AB2type Laves phase in Mg-Al-Ca alloy: Electronic structure and elastic properties [J]. Solid State Science, 2009, 11(8): 1400-1407.
[32] NYE J F. Physical properties of crystals [M]. Oxford: Clarendon Press, 1964.
[33] DUAN Y H, SUN Y, PENG M J, GUO Z Z. First principles investigation of the binary intermetallics in Pb-Mg-Al alloy: Stability, elastic properties and electronic structure [J]. Solid State Sciences, 2011, 13(2): 455-459.
[34] DAVIS L C, WHITTEN W B, DANIELSON G C. Elastic constants and calculated lattice vibration frequencies of Mg2Sn [J]. Journal of Physics and Chemistry of Solids, 1967, 28(3): 439-447.
[35] MAO P L, YU B, LIU Z, WANG F, JU Y. Mechanical, electronic and thermodynamic properties of Mg2Ca Laves phase under high pressure: A first-principles calculation [J]. Computational Materials Science, 2014, 88: 61-70.
[36] SUMER A, SMITH J F. Elastic constants of single-crystal CaMg2 [J]. Journal of Applied Physics, 1962, 33(7): 2283-2286.
[37] HILL R. The elastic behaviour of a crystalline aggregate [J]. Proceedings of the Physical Society: Section A, 1952, 65: 349-354.
[38] PUGH S F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals [J]. Philosophical Magazine A, 1954, 45: 823-843.
[39] FU C L, WANG X D, YE Y Y, HO K M. Phase stability, bonding mechanism, and elastic constants of Mo5Si3by first-principles calculation [J]. Intermetallics, 1999, 7(2): 179-184.
[40] LI X F, CHEN X R, MENG C M, JI G F. Ab initiocalculations of elastic constants and thermodynamic properties of Li2O for high temperatures and pressures [J]. Solid State Communications, 2006, 139(5): 197-200.
[41] ANDERSON O L. A simplified method for calculating the Debye temperature from elastic constants [J]. Journal of Physics and Chemistry of Solids, 1963, 24: 909-917.
[42] WACHTER P, FILZMOSER M, REBIZANT J. Electronic and elastic properties of the light actinide tellurides [J]. Physica B, 2001, 293(3-4): 199-223.
[43] HUANG Z W, ZHAO Y H, HOU H, HAN P D. Electronic structural, elastic properties and thermodynamics of Mg17Al12, Mg2Si and Al2Y phases from first-principles calculations [J]. Physica B, 2012, 407(7): 1075-1081.
[44] ZHOU D W, LIU J S, XU S H, PENG P. Thermal stability and elastic properties of Mg2X (X=Si, Ge, Sn, Pb) phases from first-principle calculations [J]. Computational Materials Science, 2012, 51(1): 409-414.
[45] MAO Ping-li, YU Bo, LIU Zheng, WANG Feng, JU Yang. Mechanical properties and electronic structures of MgCu2, Mg2Ca and MgZn2 Laves phases by first principles calculations [J]. Transactions of Nonferrous Metals Society of China, 2014, 24(9): 2920-2929.
[46] ZHAO Y, TIAN X, XUE W, GAO T. The structure, dynamical and thermodynamic properties of α-Li3N: A first-principles study [J]. Solid State Communications, 2009, 149(47-48): 2130-2134.
[47] JHA P K. Phonon spectra and vibrational mode instability of MgCNi3 [J]. Physical Review B, 2005, 72: 2145021-2145026.
王 峰,孙士杰,于 波,张 峰,毛萍莉,刘 正
沈阳工业大学 材料科学与工程学院,沈阳 110870
摘 要:通过第一性原理计算方法研究Mg-Al-Ca-Sn合金中主要强化相Mg17Al12、Al2Ca、Mg2Sn和Mg2Ca的结构稳定性、电子结构、弹性常数和热力学性质。计算所得晶格常数与实验值及文献值吻合。合金形成热和结合能计算结果表明,Al2Ca具有最强的合金形成能力和结构稳定性。通过对这些化合物的态密度、Mulliken电子占据数、金属性和差分电荷密度计算分析其结构稳定性机制。通过计算 Mg17Al12、Al2Ca、Mg2Sn和Mg2Ca 的弹性常数,推导出各相的体模量、剪切模量、弹性模量和泊松比。热力学性质计算结果表明,Al2Ca和Mg2Sn的Gibbs自由能低于Mg17Al12,即Al2Ca和Mg2Sn 的晶体结构稳定性优于Mg17Al12 相。因此,通过添加Ca和Sn元素可以提高Mg-Al系合金的热力学稳定性。
关键词:Mg-Al系合金;第一性原理计算;电子结构;弹性性质;热力学性质
(Edited by Xiang-qun LI)
Foundation item: Project (20131083) supported by the Doctoral Starting up Foundation of Liaoning Province, Clhina; Project (LT201304) supported by the Program for Liaoning Innovative Research Team in University, China; Project (2013201018) supported by the Key Technologies Research and Development Program of Liaoning Province, China
Corresponding author: Feng WANG; Tel: +86-15002424621; E-mail: wf9709@126.com
DOI: 10.1016/S1003-6326(16)64107-9

