J. Cent. South Univ. Technol. (2007)05-0618-05
DOI: 10.1007/s11771-007-0118-9 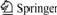
Calculation of electron structure by density function theory and
electrochemical process of surface (100) of FeS2
LI Quan(�� ȫ)1, 2, QIN Wen-qing(������)1, SUN Wei(�� ΰ)1 , QIU Guan-zhou(�����)1
(1. School of Resources Processing and Bioengineering, Central South University, Changsha 410083, China;
2. Liuzhou Huaxi Group of Guangxi, Liuzhou 515204, China)
Abstract��The electron structure of FeS2 surface (100) was computed by DFT (density function theory) and the process of electron transfer in sulfide flotation was simulated through ab-initio calculation. The results show that the interaction between xanthate and FeS2 is controlled by the energy of valence band. The products and degree of the reaction depend on the density of state of valence band and concentration of positive hole in valence band. Interaction between xanthate and pyrite can be changed by modifying the election structure of the surface of pyrite. Xanthate is adsorbed on the surface of intrinsic pyrite. But the amount of xanthate adsorbed on the surface of the pyrite with sulfur vacancy is more than that on the surface of the intrinsic pyrite due to the higher electron and vacancy density. Xanthate is not adsorbed on the surface of pyrite with Fe vacancy because of its high Fermi energy.
Key words: pyrite; electrochemiscal process; flotation; ab-initio calculation
1 Introduction
Pyrite(FeS2) is the most abundant sulfide mineral and an important mineral material. The surface chemistry and surface reactivity of pyrite have been extensively studied, particularly their importance in the processing of sulfide ores[1-4]. The flotation of sulfides is an electrochemical process, and adsorption of collectors on the surface of mineral results from the electrons transfer between mineral surface and oxidation-reduction composition in pulp[5]. According to electrochemical principles and semiconductor energy band theories, it is known that the kind of electron transfer process is decided by the electronic structure of mineral surface and oxidation-reduction activity of reagent[6]. In the field of mineral processing, researches are now carried out mainly by using electrochemical testing to probe into oxidation-reduction characteristics of minerals, or using spectrum way for token, and deeper analysis to surface of mineral from quantum chemistry is scarcely seen[7-9]. The bulk and surface properties of FeS2 were investigated in Refs.[10-15]. By using various kinds of density functional theories(DFTs), it is found that such methods are capable of producing calculated bulk properties such as lattice constants, which agree well with those of experiment, and provide a reference for the study of pyrite surfaces[10-15].
In this study, quantum chemistry calculation was carried out to mineral surface by using DFT as tools and pyrites as samples, and then, according to the electronic structure characteristics of collectors, the reaction between pyrites and collectors in molecule layer was investigated to find the ultimate reason for pyrites flotation. DFT method was used to model FeS2 surface (100) and its interactions with xanthate. The electronic structure of FeS2 surface (100) was calculated to study the surface properties and xanthate adsorption behavior on the surface.
2 Ciculation model
From mineralogy theory, it is known that the cleavage surface of pyrites is surface (100), and the model of FeS2 surface (100) is shown in Fig.1. The calculation model of this research was constructed in accordance with surface (100). When the model was constructed, cleavage surface (100) was gotten from the constructed pyrite crystal model with CLEAVAGE tool in C2 software package first, and then three-dimension disposition to the cleavage surface was carried out with CRYSTAL BUILDER tool and the preceding model was established finally. In the preceding model, a=b=0.543 nm, c=2 nm. The length of bond Fe��S and S��S is 0.227 and 0.215 nm, respectively, and the angle of Fe��S��Fe is 115?. All the calculation (including model construction) in this research was processed in C2 software of SGI workstation. Calculation about the mineral surface was carried out with CASTEP tool and calculation about molecule relation energy and exchange energy was carried out with potential energy function of PEDREW-WANG. The pattern of the calculation is water solution, and semidiameter of hydrated membrane is, approximately, the depth of single molecule layer adsorption, namely 0.15 nm.
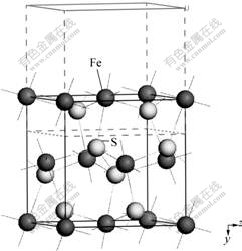
Fig.1 Model of cleavage surface (100) of FeS2
It is considered commonly that oxidization of xanthate in the pyrite flotation takes place in ion form, so xanthate model in this research was constructed in ion form. To simulate a water solution system, xanthate carried one electric charge was laid in a 1 nm3 ��water tank��, and in the ��water tank��, there are 34 water molecules scattered stochastically, after that dynamic stimulation with 1��107 steps was proceeded in the way of molecule force field (MM+), the structure was stable. The model is shown in Fig.2. From Fig.2, water molecule gathered to hydrophilic radical can be seen. It makes clear that construction of the model is suitable to
the fact. Finally, the single point energy of xanthate ion(X-) was calculated with Gaussian method, and its basis set function was 6-31G.
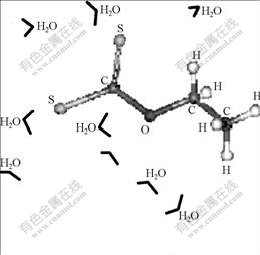
Fig.2 Model of xanthate anion in water tank after geometric optimization
3 Results and analysis
3.1 Analysis of energy band structure on pyrite surface
Fig.3 shows energy band structure of pyrite (100) surface and density of state(DOS) of area nearby conducted band and valence band, which were calculated with CASTEP tool. Fig.3 shows that the bottom of pyrite��s conducted band lies in range from �C0.400 to -0.135 eV, and its width is 0.27 eV; the top of the valence band lies in range from -1.735 to -1.600 eV, and its width is 0.13 eV, the width of the forbidden band is 1.2 eV, close to that of latent pyrite (0.9 eV). This also shows that pyrite is a typical semiconductor (width of the forbidden band is small)[16]. Fermi energy level of pyrite surface is approximately equal to the average value of energy level of conducted band��s bottom, and valence��s top, namely �C0.1 eV. The analysis to the distribution of DOS shows that there is a pinnacle near the conducted band and partial DOS analysis indicates that DOS nearby the conducted band is contributed mainly to 3d orbit of Fe, whereas that nearby the valence band is contributed mainly to 3p orbit of S.
Fig.3 Energy band structure of cleavage surface (100) of FeS2 and partial DOS analysis
3.2 Configuration of xanthate and orbit analysis
Configuration parameters of xanthate ion, electric charge carried by each atom is listed in Table 1 and orbit energy level distribution of xanthate after Gaussian calculation is shown in Fig.4. It can be seen that the centers of negative electric charge of xanthate lie on the two sulfur atoms, electric charges carried are -0.663 and -0.668 eV, respectively. There is little difference between them. Moreover, the two atoms lie in the same plane with carbon atom and oxygen atom, which suggests that there is conjugate effect on them. Fig.4 also shows that the highest occupied molecule orbit (HOMO) energy level and the lowest unoccupied molecule orbit (LUMO) energy level of xanthate is �C1.891 and 1.846 eV, respectively, its Fermi energy level is approximately the average value of the two orbits energy level, namely �C0.023 eV, which accords with the reports in literatures.
Table 1 Electric charge carried by each atom in xanthate

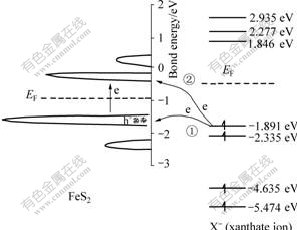
Fig.4 Demonstration of electron transfer between FeS2 and xanthate ion
3.3 Model of electron transfer of xanthate on surface of pyrite
According to energy band theory, it can be found that, to the ��eigen semiconductors�� that have not any impurities or disfigurement, at 273 K, all the energy levels of valence band are filled with electrons, while conducted band is empty and there are forbidden bands between them where the quanta state electrons do not exist. But if the temperature is higher than 273 K, it is possible for electrons to obtain energy higher than the width of the forbidden band because of heat blaze, the semiconductor material will have definite capability of electric conduction. After being blazed, electrons of the valence band will enter into the conducted band, then vacant sites will be left in the valence band, which is called ��cavity��. Thereby, there are two kinds of current carriers in semiconductor: free electrons in conducted band and cavities in valence band.
At the interface of semiconductor/solution, two kinds of electric charge reaction, namely electrons transfer in conducted band and cavities transfer in valence band, are possible:
R-e O (1)
O (1)
R+h+O (2)
Formulas above show two adverse processes: current carriers in the electrode are captured by ions in the solution and ions in the solution are poured into the electrode. If the reaction is sorted according to the type of current carriers that attends the reaction in the semiconductor, there are exchange reaction. The conducted exchange reaction of semiconductor of N type is donor exchange reaction (Eqn.(1)), whereas exchange reaction concerning to cavities in valence band is acceptor exchange reaction (Eqn.(2)). If 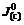 and
and  stand for exchange current density of conducted band and valence band, respectively, it is easy to obtain the similar formula.
stand for exchange current density of conducted band and valence band, respectively, it is easy to obtain the similar formula.
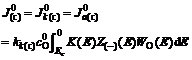
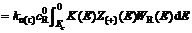 (3)
(3)
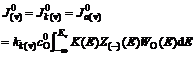
 (4)
(4)
where 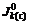 ,
,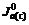 ,
, and
and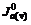 stand for current densities of cathode and anode in the conducted band and the valence band in balance. Z(-)(E) and Z(+)(E) are density functions of energy levels occupied and unoccupied by electrons in the semiconductor, respectively. WO(E) and WR(E) stand for distribution functions of oxidation state and deoxidation state in the solution.
stand for current densities of cathode and anode in the conducted band and the valence band in balance. Z(-)(E) and Z(+)(E) are density functions of energy levels occupied and unoccupied by electrons in the semiconductor, respectively. WO(E) and WR(E) stand for distribution functions of oxidation state and deoxidation state in the solution. 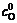 and
and 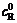 stand for respectively the balance concentrations of oxidation and deoxidation state in the solution. K(E) is probability factor (frequency factor).
stand for respectively the balance concentrations of oxidation and deoxidation state in the solution. K(E) is probability factor (frequency factor).
Since electron transfer takes place mainly in a narrow energy field nearby band margin of the semi- conductor, only electrons at energy levels that are not so far from the band margin than KT scope have the largest exchange velocities. Then the diversified current densities can be expressed by the following formulas approximatively:

 (5)
(5)

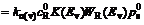 (6)
(6)
where  and
and 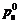 stand for electrons�� density and cavities�� density of the semiconductor��s surface. Overall exchange current density in the system when it reaches balance is the sum of exchange current of conducted band and valence band.
stand for electrons�� density and cavities�� density of the semiconductor��s surface. Overall exchange current density in the system when it reaches balance is the sum of exchange current of conducted band and valence band.
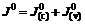 (7)
(7)
Because the energy band��s position of the interface��s two sides is asymmetry usually only one kind of exchange current is main in common situation. To semiconductors that have different widths of forbidden band, the corresponding energy levels of the interface��s two sides may be changed because of the change of band margin��s position, thus nature of the reaction will be changed too. Taking the interactions between ions of pyrites and xanthates as an example, Fig.4 shows the interactions of energy levels of them. The left in Fig.4 is DOS nearby conducted band and valence band of the pyrite; the right of it is front orbit energy band distribution of the xanthate. Because of the temperature is higher than 273 K, thermo-transition of electrons on the pyrite��s surface will take place, resulting in a certain certain quantity of cavities and electrons at the top of valence band and the bottom of conducted band.
It is reported that xanthate(X) will react on the surface of pyrites:
2X--2e X2 (8)
X2 (8)
Fig.4 shows that Fermi energy level of xanthate ions is higher than that of the pyrite surface, so electron will transfer from xanthate to the pyrites�� surface, therefore reaction above is approved. There are two kinds of transfer (Fig.4), according to energy level matching principles, electrons are apt to transfer in the first way. That is to say, the valence band controls electrons�� transfer then. Eqn.(6) shows that valence band controlled current density is decided by cavity concentration of the semiconductor surface , energy density functions of levels occupied and unoccupied by electrons in the semiconductor Z(-)(Ev) and Z(+)(Ev), distribution functions of oxidation state and deoxidation state in the solution WO(E) and WR(E), balance concentrations of oxidation and deoxidation state in the solution and 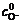 To xanthate in a certain concentration, the other parameters are constants except for , and the reaction is mainly decided by the cavity concentration of the semiconductor��s surface. That is to say, exchange current density in the pyrite and xanthate system gives the first place to valence band exchange current density.
To xanthate in a certain concentration, the other parameters are constants except for , and the reaction is mainly decided by the cavity concentration of the semiconductor��s surface. That is to say, exchange current density in the pyrite and xanthate system gives the first place to valence band exchange current density.
It can be inferred from above analysis that, if some impurities are mixed into pyrite, or certain kinds of disfigurements are made by certain special technics (grinding etc.), there will be a low object energy level in pyrite, then the transition of electrons becomes easier, cavity density in valence state becomes larger, which means that the reaction between pyrite and xanthate will be enhanced.
3.4 Illustration of electrons transfer of xanthate on surface of pyrite
Fig.5 shows the valence electrons�� distribution of front obits between pyrites and xanthate after Gaussian calculation. It shows clearly that electrons are apt to transfer from xanthate to pyrite, which is in accordance with forecast results of energy level theory.
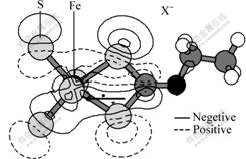
Fig.5 Contour plot of electron transfer between FeS2 and X-
4 Conclusions
1) Pyrite is a nice semiconductor, the forbidden band on its surface is only 1.2 eV.
2) Electrons transfer between pyrite and xanthate is the reaction controlled by valence band.
3) Interaction between xanthate and pyrite can be controlled through modifying the electron structure of the surface of pyrite.
References
[1] RAICHUR A M, WANG X H, PAREKH B K. Quantifying pyrite surface oxidation kinetics by contact angle measurements[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2000, 167(3): 245-251.
[2] BALTRUS J P, DIEHL J R. Surface spectroscopic studies of factors influencing xanthate adsorption on coal pyrite surfaces[J] . Fuel and Energy Abstracts, 1997, 38(4): 212-216.
[3] BOON M, HEIJNEN J J. Solid-liquid mass transfer limitation of ferrous iron in the chemical oxidation of FeS2 at high redox potential[J]. Hydrometallurgy, 2001, 62(1): 57-66.
[4] LAAJALEHTO K, LEPPINEN J, KARTIO I, et al. XPS and FTIR study of the influence of electrode potential on activation of pyrite by copper or lead[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 1999, 154(1/2): 193-199.
[5] KELSALL G H, YIN Q, VAUGHAN D J, et al. Electrochemical oxidation of pyrite (FeS2) in aqueous electrolytes[J]. Journal of Electroanalytical Chemistry, 1999, 471(2): 116-125.
[6] BUSWELL A M, NICOL M J. Some aspects of applied electrochemistry of the flotation of pyrrhotite[J]. Journal of Applied Electrochemistry, 2002, 32: 1321-1329.
[7] QIN Wen-qing, QIU Guan-zhou, WANG Dian-zuo. Galvanic interaction of contacting sulfide particle and its effect on flotation[J]. Trans Nonferrous Met Soc China, 1998, 8(4): 457-462.
[8] QIN Wen-qing, QIU Guan-zhou, HU Yue-hua. Dynamics of electrodeposition of tetraethylthioram disulphide(TETD) on pyrite surface[J]. Journal of Central South University of Technology, 2001, 8(3): 164-168.
[9] BUSWELL A M, BRADSHAW D J, HARRIS P J. The use of electrochemical measurements in the flotation of a platinum group minerals (PGM) bearing ore[J]. Minerals Engineering, 2002, 15(6): 395-404.
[10] HUNG A, MUSCAT J. Density functional theory studies of pyrite FeS2 (100) and (110) surfaces[J]. Surface Science, 2002, 513(3): 511-524.
[11] HUNG A, MUSCAT J. Density functional theory studies of pyrite FeS2 (111) and (210) surfaces[J]. Surface Science, 2002, 520(1/2): 111-119.
[12] OPAHLE I, KOEPERNIK K, ESCHRIG H. Full potential band structure calculation of iron pyrite[J]. Computational Materials Science, 2000, 17(2/4): 206-210.
[13] EDELBRO R, SANDSTR J M A, PAUL J. Full potential calculations on the electron band structures of sphalerite pyrite and chalcopyrite[J]. Applied Surface Science, 2003, 206(1/4): 300-313.
[14] MUSCAT J, HUNG A, RUSSO S, et al. First principles studies of the structural and electronic properties of pyrite FeS2[J]. Physical Review, 2002, 65(1): 54-57.
[15] SUN Wei, HU Yue-hua, QIU Guan-zhou. Oxygen adsorption on pyrite (100) surface by density functional theory[J]. Journal of Central South University of Technology, 2002, 11(4): 385-390.
[16] ALTERMATT P P, KIESEWETTER T. Specifying targets of future research in photovoltaic devices containing pyrite (FeS2) by numerical modeling[J]. Solar Energy Materials and Solar Cells, 2002, 71(2): 181-195.
Foundation item: Project(20047) supported by the Foundation of National Excellent Doctoral Dissertation of China; Project(50204013��supported by the National Natural Science Foundation of China
Received date: 2007-01-20; Accepted date: 2007-03-23
Corresponding author: QIN Wen-qing, Professor, PhD; Tel: +86-731-8879622; E-mail: QWQ@mail.csu.edu.cn
(Edited by CHEN Wei-ping)

