���±�ţ�1004-0609(2008)12-2233-12
��ĥ������Fe�Ͻ���MgH2 ��ϵ���ܵĻ���
�ܵ���1���� ��1, 2���� ƽ2������ˮ2
(1. ���ϴ�ѧ ���������Ƚ������������ص�ʵ���ң���ɳ 410082��
2. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ����ɳ 410082)
ժ Ҫ�������ܶȷ������۵ĵ�һԭ��������ͨ��������ࡢ��λȱ��Mg(0001)�������������(H2)ǰ���Լ�Fe�Ͻ�þ�⻯����(MgH2)��ϵ����������ӽṹ������ĥ������Fe�Ͻ���MgH2��ϵ���ܵ�ԭ����г���̽�֡���������������Mg������ȣ�������ĥ�ı���Mg�����ı���ṹ��ʹMg��������϶�ȱ�ݣ���ȱ�ݵĴ�����ǿ��H2��������������������Mg������H2ת�Ƶĵ�������࣬�����ϵ��ĥ����нϺõ��������ܣ�����Fe�Ͻ�MgH2��ϵ�У�Fe������MgH2���γ�(MgFe)H2������ͺϽ��γ�����Mg2FeH6�࣬MgH2ϵ�ṹ�ȶ��Ծ����ͣ���Ӧ��ϵ����������ǿ���������ӽṹ���֣���λȱ������H2������Mg���棬��Mg(0001)�������ϲ���H2ֱ�Ӳ����������õĽ���ԭ���ڷ����ܼ�(EF)����s����ijɼ�������������أ���Fe�Ͻ�MgH2��ϵ�У���Ͻ�Ԫ��Fe���ڵ�Hԭ���γɿ�λ���Ѷ����ӣ�Hԭ�ӽ����ͷţ���Mg���ڵ�Hԭ���γɿ�λ���Ѷȼ��٣�Hԭ�������ͷţ�Fe�Ͻ���Mg-H֮����ڽ����ijɼ����ã���ˣ�MgH2��ϵ�Ľ������ܵõ���ߡ�
�ؼ��ʣ�MgH2��Mg(0001)���棻�������γ���
��ͼ����ţ�TG 146.2 ���ױ�ʶ�룺 A
Mechanism of improved properties of MgH2 systems with ball milling and iron addition
ZHOU Dian-wu1, ZHANG Jian1, 2, PENG Ping2, LIU Jin-shui2
(1.State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, Hunan University, Changsha 410082, China;
2. School of Materials Science and Engineering, Hunan University, Changsha 410082, China)
Abstract: The energy and electronic structure of the adsorption of H2 on the clean, vacancy defective Mg (0001) surfaces and iron alloying magnesium hydride were calculated by using a first-principles plane-wave pseudopotential method, the mechanism of improved properties on MgH2 systems were analyzed with milling and iron addition. The results show that the vacancy defect system benefits enhancing the physical sorption between Mg surface and H2 compared with the clean Mg (0001) surface, because the quantity of the charges transferring from Mg surface to H2 adsorbed may be increased significantly, the H2 adsorption properties are improved after ball milling. The structure stability of the alloying system is reduced when a little iron dissolves into the magnesium hydride, and is further reduced when the iron additions form Mg2FeH6 compound. The analyses of electronic structures shows that the catalytic reactivities for H2 adsorption of the different surfaces are dependent on the numbers of s orbital bonding electrons around Fermi level for the uppermost layer metal atoms which interact directly with H2. It is easy to form vacancy for hydrogen atoms next to iron atom, which indicates that hydrogen atom cannot be escaped, but it is difficult to from vacancy for hydrogen atoms next to magnesium atom, which indicates that hydrogen atom can be escaped. Hence it is thought that the change of dehydrogenating properties of MgH2 with or without a little iron addition attributes to the weakened bonding between magnesium and hydrogen.
Key words: magnesium hydride; Mg(0001) surface; adsorption; formation heat
���Ż�ʯ��Դ���ݽߺͻ�����Ⱦ���������أ�����һ�������ʯ��Դ����������Դ����һ�����ȵĿ��⡣������Ϊ�ྻ����Դ�����Ϊ����ע�Ľ��㡣����������������ü�������Ĺؼ�����֮һ���������������ڶ��о���ԱΪ��չ�����ܴ�����Ͽ�չ�˹㷺���о�����������������ܵ�������Ҫ�����Ǵ��������������ѧ��þ�⻯����(MgH2)���۴�����(7.6%����������)���ڹ�����Դ����(IEA)ȷ��δ�����ʹ�����ϵı�5.0%������Ϊ������Ͻ���Ӧ������ջ�����Ȼ������ϵ���������ܲ��������ʵ��Ӧ�á�Ϊ�����ϲ�������ѧ�����Dz�����ĥ���������ͳ�ĸ�������������������ĥ�ı���Mg�����ı���ṹ��ʹ��������϶�ȱ�ݣ������ĥ���Ʊ���Mg�������ȴ�ͳþ�۾��и���������ѧ[1?2]��Ϊ����MgH2�ϲ�Ľ����ѧ��Ŀǰ�������������������ϵ�ڼ���������������й��ɽ���Fe�Ͻ�����֤����һ����Ч������ʵ�鷢�֣���MgH2 ����Fe�Ͻ���Fe��MgH2Ħ����Ϊ8?100[3]��������Ϊ1?10 [4]ʱ����һ���¶Ⱥ�ѹ���£����������������ĥ20 h����������ղ���Ϊ��-MgH2+Fe������ĥ�ﵽ6 h��[5]���������µķ�����Mg2FeH6��HIGHTOWER��[6]�ڲ���Fe�Ͻ�MgH2�Ʊ�Mg2FeH6�Ĺ����з��֣�Fe��Mg��������ܣ���HUOT��[5]����Mg2FeH6���ɵ�ͬʱ���Ͻ���ϵ�б�����δ��Ӧ��Mg����MgH2��ȴ��ʧ�ˡ�����������ߺ������ֵ�[7]���õ�һԭ�������������ĥ���ϵķ������ڲ���ʵ��XRD��ȷ����ϵ����ɵĻ����Ͻ�������ģ�ͣ��������ۼ��㷽��������Mg2FeH6���γɹ��̽��г����о�������ʵ��DSC���ԵĺϽ��γ��������ۼ���ĺϽ��γ����ڱ仯����һ�µ�ǰ���£�ͨ��ʵ������Ʊ�������Mg2FeH6����Ͻ𡣱���������ʵ��DSC����ʱ���֣�����ȡ����ĩ����ʾ����1�����ȷ壬��ʹMg2FeH6��MgH2����棬ҲΪ1�����ȷ壻������ĥ��ĩ���ڲ�����300 ��������½��⣬�ɴ˿ɼ�����MgH2����Fe�Ͻ��Ը�����ϵ�������ܡ�
ͨ��Mg�����ѧ�������ṹ������أ�Ϊ�����Ƕ�H2����Mg���濪չ�˴�����������SPLUMMER��[8]����������ʧ��(EELS)������ѧ�Ѹ�����(TDS)�о���H2���ӡ�Hԭ����Mg֮�������ã�����H2��Mg�����������Ҫ�������Mg������ڽ�ǿ�Ļ�ѧ����Hԭ�ӵı����⻯��㣻N?RSKOV��[9]���ô�ͷ�㷨�о���H2��Mg(0001)���������������H2��Mg�����������ʽΪ��ѧ������VEGGE��[10]ȴ����Mg��������H2�Ĺ��̽�Ϊ��������������Fe�Ͻ���MgH2�������ܵ������о���SONG��[11]����ȫ������ƽ�沨(FLAPW)��������Ϊ�Ͻ�Ԫ��Fe����MgH2��ϵ�������ܵ���Ҫԭ�����ڣ�Fe�ļ�����������ϵ�����ܼ�����Mg��H�ɼ��������ı仯��
����������Ϊ���MgH2��ϵ�ϲ���������ܣ�������Ȼ������ĥ�����ͳ�ĸ���������չ��һ����ʵ���о�����H2��Mg���������Ҳ����һЩ����̽�֣��������ķ�ʽ�۵�ȴ��һ�£����Ҷ�H2�ڸ���(���λȱ��)Mg���������������о����ټ������ױ���������������ĥ�Ʊ�Mg2FeH6���ڽ�����Ψһ�⻯��MgH2[7]����ܿ�������ĥMg��������϶�ȱ�ݣ�����H2�������ױ�Mg��������������Mg��H2��Ӧ�γ�MgH2����ȱ���������ݡ�����Mg2FeH6����ṹĿǰȱ�����������ݣ�SONG��[11]û�бȽ�MgH2����ͬFe����(MgFe)H2������Ľṹ�ȶ����Լ�Mg2FeH6��MgH2��ϵ�������ܵ�Ӱ�죬Ϊ�ˣ��������߲��û����ܶȷ������۵ĵ�һԭ�����㷽����ͨ��������ࡢ��λȱ��Mg(0001)�������������(H2)ǰ���Լ�Fe�Ͻ�MgH2��ϵ����������ӽṹ��̽����ĥ������Fe�Ͻ���MgH2��ϵ���������ܵ��ۻ���������Ϊ�������þ����������ṩ����ָ����
1 ���㷽��
Ϊ�о���ĥ�ı�Mg��������ṹ��ʹMg��������϶�ȱ�ݣ�����H2��Mg������������������MgH2�������ܵĻ��������о��м�����ϵ����������ӽṹʱ�����û����ܶȷ������۵�Dmol 4.1�����ӽ��������ܺ�������GGA���Ƶ�PW91��ʽ[12]���ƺ���ȡȫ����λ�ƣ����Ӳ���������DND�� ��[13?14]��������K��������ȡ4��2��1�Ϻ�������ϵ�Ż�ʱ���侫������Ϊ������ ��2.0��10?5 Ha��Ӧ����0.004 Ha��λ�ơ�0.005 ?���о�Fe�Ͻ���MgH2��ϵ�������ܣ����о��м�����ϵ����������ӽṹʱ��ʹ�õ���CASTEP(Cambridge Serial Total Energy Package)���ܼ���������[15?16]�����������ܲ��ù����ݶȽ���(GGA)�е�Perdew-Burke-Ernzerhof��ʽ[17?18], ���������ܵļ�������С���Ŀ��ٸ���Ҷ�任(Fast-Tourier-Transform, FFT)�����Ͻ��У����ö������������г�ԥ�ij���(Ultrasoft)����[19]��Ϊƽ�沨����[20]��������Ǣ����(SCF)�������м��㣬����ʱ�����ý��Broyden-Flecher-Goldfarb-Shanno(BFGS)�����ݶȷ���[21]��Pulay�ܶȻ�Ϸ���[22]�������ӳ�ԥ���ȶ�ģ�͵ľ���ṹ������ȫ�ļ����Ż�����������ǵľ������ȶ��ṹ���ټ����Ż���ģ�͵ĵ����ܡ��Ż�����ʱ����ϵ������������ֵȡ2.0��10?5 eV/atom, ÿ��ԭ���ϵ�������0.05 eV/?, ����ƫ��С��2.0��10?3 ?��Ӧ��ƫ��С��0.1 GPa��ƽ�沨չ���Ľ�ֹ����EcutΪ310.0 eV��FFT����Ϊ24��24��80�����õ�K�ռ�Ϊ0.50 nm?1������Monkhorst-Pack�㷨���õ�K������Ϊ(4, 4, 1)�������ڵ��ռ���С�ͨ�������Եļ��飬������Щ�趨�������Ա��ּ�������Ҫ�ľ��ȡ�
2 �����������
2.1 Mg(0001)����ģ��
Ϊ��������̽����ĥ����MgH2��ϵ�������ܵ��ۻ����������Ϻò��ԣ����о��й�����5��Mg(0001)������ģ�ͣ���ṹʾ��ͼ��ͼ1(a)��ʾ�����DZ߽�ЧӦ��������ã��䳬����С��Ϊp(2��3)��Ϊ����㾧֮�������ţ���ղ���ȡ20 ?����λȱ��Mg(0001)����ģ��ʾ��ͼ��ͼ1(b)��ʾ������ģ����ϵ�Ż�ʱ�������������Mgԭ�ӡ�H2���ӻ�Hԭ���������ɳ�ԥ������3��Mgԭ�ӱ��̶���

ͼ1 5��Mg(0001)����ģ��ʾ��ͼ
Fig.1 Schematic diagram of models of Mg(0001) surfaces: (a) Clean; (b) vacancy defective sufaces
2.2 MgH2�ľ���ṹ
MgH2�ľ���ṹʾ��ͼ��ͼ2(a)��ʾ�����ɾ�����a��c�������ռ�ȺΪP42/mnm(NO.136)�������и�ԭ������Ϊ��+2Mg(0, 0, 0)��+4H(0.304, 0.304, 0)[23]������ʵ�ʲ�����a��cһ�㲻�������ܶȷ������㣬�����ܶȷ������ۼ����a��c���뾭����Ӧ�ļ��㷽�������Ż��������������ܺ��������ݵ�Ʒ�ʶ����Ż�a��c�о��������ã���ˣ�������ʹ��Ż�a��c��ʼ����MgH2����ṹ�ļ����У��Խ��������ܹ����ݶȽ���(GGA)��3����ʽ(PBE��PRB��EPW91)�;ֲ��ܶȽ���(LDA)��CA-PZ��ʽ�����������ֲ�ͬ�����ƣ�Ultrasoft��Norm-conserving���ֱ�����˾�����a��c��a/c��ֵ������ʵ��ֵ�Լ����˵ļ����������˱Ƚϣ�������ڱ�1��
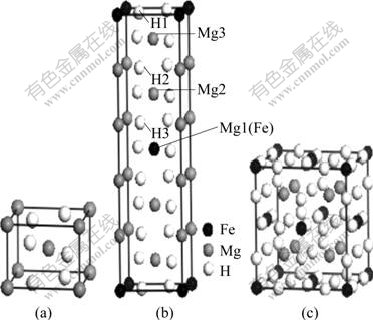
ͼ2 ��λȱ��Mg(0001)����ģ��MgH2��Mg2FeH6�ľ���ṹʾ��ͼ
Fig.2 Schematic diagram of crystal structures for MgH2 and Mg2FeH6: (a) MgH2; (b) Five-times unit of MgH2; (c) Mg2FeH6
��1 MgH2������(a, c, a/c)�ıȽ�
Table 1 Lattice parameters(a, c, a/c)of MgH2 by GGA, LDA, experiment and calculation
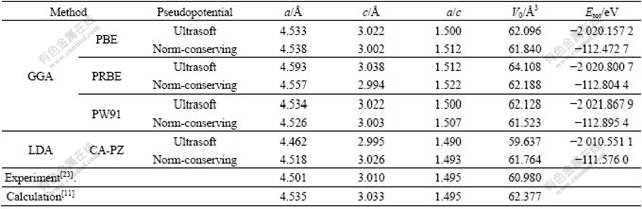
�ӱ�1�ɼ������������������ܺ����Ƶļ������Ƚϣ����������ܲ��ù����ݶȽ���(GGA)��PBE��ʽ������ȡ���ռ�����ij���(Ultrasoft)���ƣ�MgH2��ƽ�⾧����a��c�ֱ�Ϊ4.533��3.022 ?����ʵ��ֵ[23] (a=4.501 ?��c=3.010 ?)��ӽ�������SONG��[11]�ļ�����(a=4.535 ?��c=3.033 ?)���ϵúܺã����һ����֤�˼��㷽����ѡ�������ܺ����Ƶ�ȷ�ԡ�
2.3 Fe�Ͻ�MgH2��ϵ����ģ�ͺ�Mg2FeH6����ṹ
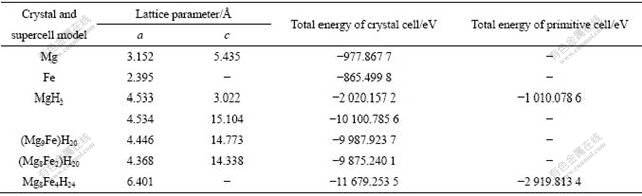
Ϊ�о�Fe�Ͻ���MgH2��ϵ�������ܵ��ۻ������ٶ�Fe������MgH2���γ�(MgFe)H2�����壬�䳬��ģ�ͽṹ��ͼ2(b)��ʾ�����ǵ���Hԭ����ȣ�Feԭ����Mgԭ�Ӵ�С���ӽ�����1��Feԭ�����ͼ2(b)ģ�����ĵ�1��Mg1ԭ�ӣ��õ�(Mg9Fe)H20��������ʱ��ӦFe��MgH2�еĹ���Ũ��(Ħ������)Ϊ5.0%����2��Feԭ��������ĺ�8�������1��Mg1ԭ�ӣ��õ�(Mg8Fe2)H20��������ʱ��ӦFe�Ĺ���Ħ������Ϊ10.0%������MgH2����ģ�͵�ƽ�⾧�������2���С��ɱ�2��֪��Fe�Ͻ�ƽ�⾧����a��c�����١�
��2 ����ͳ���ģ�͵�ƽ�⾧����(a)��(c)�Լ�������
Table 2 Equilibrium lattice constant (a), (c) and total energies of crystal and supercell model
Mg2FeH6�ľ����ṹ(Mg8Fe4H24)��ͼ2(c)��ʾ��Ϊ��CaF2�ṹ�����ɾ�����a�������ռ�ȺΪFm3m (NO.225)�������и�ԭ������Ϊ��+8Mg(0.25, 0.25, 0.25)��+4Fe(0, 0, 0)��+24H(x, 0, 0)��xΪ(0~0.240)[24]������ʵ��δ����Mg2FeH6�����ṹ(Mg8Fe4H24)��Hԭ�ӵ�λ�ã�Ϊ�ˣ�ѡ��ʵ��ֵ������ͬ�ľ������Ͳ�ͬ��xֵ������һϵ�еļ��㣬����Mg2FeH6�����ƽ�⾧���� (6.401?)��ʵ��ֵ (6.443?[24]��6.444 ?[5]��6.442 ?[25]��6.429 ?[26])���ϵýϺã�ͬʱ��Ӧx=0.240ʱ��Mg2FeH6������������(?11 679.253 5 eV)Ϊ���ֵ������������Խ�ͣ���Ӧ�����ṹԽ�ȶ���Ϊ��ȷ��Mg2FeH6�����ṹ��Hԭ�ӵ�����Ϊ(0.240, 0, 0)��
2.4 H2��Mg(0001)���������
Ϊ̽����ĥMg�Ƿ�����H2��Mg�����������Ӷ�����MgH2��ϵ���������ܣ����о��м�����H2����ࡢ��λȱ��Mg(0001)����������ܡ�����ʱ��H2�ij�ʼ����λ������Mg(0001)����������H2������λ�����Ϸ�[27]�����Ҿ�Mg����������߶Ⱦ�Ϊ3 ?, ��ͼ3��ʾ��Ϊ���ڱȽϣ�Ҳ�����˱����H2��ƽ�������ܡ�������Eads(H2)��ƽ�������� (H2)�ļ��㹫ʽ����[28]��
(H2)�ļ��㹫ʽ����[28]��

���������ڱ�3��

ͼ3 H2��Mg(0001)����ij�ʼ����λ
Fig.3 Initial positions of H2 on Mg (0001) surfaces during process of H2 adsorption: (a) Clean surface; (b) Vacancy defective surface
��3 H2����ࡢ��λȱ��Mg(0001)�����ȶ�������������(Eads(H2))��ƽ�������� (H2)
(H2)
Table 3 Adsorption energy (Eads(H2)) and average adsorption energy ((H2)) of clean and vacancy defective Mg (0001) surface during H2 adsorption process

�ɱ�3�ɼ��������ܾ�Ϊ��ֵ���������������ͷ�������H2��Mg(0001)������������Խ��С������ܱ���ԭ�����༰��Ŀ��Ӱ�죬�������Mg(0001)�����H2��������Eads(H2) (0.025 0 Ha)���Ը��ڿ�λȱ�ݵĶ�Ӧֵ(0.023 1 Ha)��Ȼ��������������H2ֱ�Ӳ����������õĽ���ԭ��ʱ�����ֿ�λȱ��Mg(0001)�����H2��ƽ��������(0.004 6 Ha/atom)�������Mg(0001)����(0.004 2 Ha/atom)�����������λȱ�ݵĴ���������ǿMg�����H2�����������������ĥ����H2��Mg���������������ٽ�Mg��H2��Ӧ�γ�MgH2�������ĥ�Ʊ�Mg2FeH6���ڽ�����Ψһ�⻯��MgH2[7]��������ĥ�ı���þ�����ı���ṹ��ʹ��������϶�ȱ�ݣ������ĥ���Ʊ���þ�������ȴ�ͳþ�۾��и���������ѧ[1?2]��
ͼ4��ʾΪH2�ȶ�������Mg(0001)����ĵ����ܶ�ͼ����ͼ4(a)�ɼ���H2�ȶ����������Mg(0001)����ʱ��H2����������汣��ƽ�У��������������У�H2ֻ���ڴ�ֱ�ڱ��淽������ƽ�ƣ���ʱ��H2��Mg����伸���������ص�������ϵ��Hԭ������������Mgԭ�Ӽ��Ϊ3.937 8 ?��Զ����H?(1.54 ?)��Mg2+(0.66 ?)�����Ӱ뾶֮��2.20 ?������H2�����Mg(0001)����������������������H2����Ϊ0.748 9 ?��������H2����(0.747 6 ?)��ȣ��仯��С��H2�ȶ������ڿ�λȱ��Mg(0001)����ʱ������Mgԭ�ӿ�λ�Ĵ��ڣ�H2����Ťת�������λ��������������������һ���н�(��ͼ4(b))����ʱ��H2��Mg�����������������ζ��ԣ����нϴ�̶ȵĵ������ص���Hԭ������������Mgԭ�Ӽ��Ϊ3.601 5 ?�����Դ���H+ (1.54 ?)��Mg2+(0.66 ?)�����Ӱ뾶֮��2.2 ?���������Mg(0001)������ȣ������С��0.346 3 ?�����ͬʱ���������H2����Ҳ�쳤Ϊ0.755 6 ?������H��H���õ�һ���̶ȵ����������ƺ�Ԥʾ�ţ�Mg�����λȱ�ݵĴ��ڣ���ǿ��H2��þ�������������������

ͼ4 H2����ࡢȱ��Mg(0001)�����ȶ��������͵ĵ����ܶ�
Fig.4 Electron density contours of Mg (0001) surfaces during process of H2 adsorption: (a) Clean surface; (b) Vacancy defective surface
��4��һ��������Mg(0001)�����ȶ��������͵�Mullikenԭ�Ӳ��ӡ�����H2�ȶ����������Mg(0001)����ʱ��������Mg������H2�ϲ���������ת�ƣ��������H2���м������ĸ����(?0.015)��������Mgԭ������������������(0.035)����H2�ȶ������ڿ�λȱ��Mg(0001)����ʱ������������ȣ�H2��Mg����õ��ĵ�����ȴ��������(?0.042)��
��4 H2����༰ȱ��Mg(0001)�����ȶ��������͵�Mulliken���ӷ���
Table 4 Mulliken population of clean and vacancy defective Mg (0001) surfaces during H2 adsorption process

2.5��Fe�Ͻ�MgH2��ϵ�Ľ�������
ͨ��MgH2��ϵ�������ѧ����p��c��T���߱�ʾ��һ���¶�(T)��ѹ��(p)�£��Ͻ��γ���?H�ĸߵ;����Ž���ƽ̨ѹ�Ĵ�С��һ����ԣ�?HΪ��ֵ����ֵԽ��������ƽ̨ѹԽ�ͣ�MgH2��ϵ����Խ�ѣ���?H�ľ���ֵԽС��Խ������[29?30]�����ڴˣ����о�����MgH2��ϵ����������þ�Լ�������������IJ�ֵ����ʾ����þ�й��ܻ��γ��⻯��ĺϽ��γ��ȣ����㹫ʽ���£�

Fe��MgH2�й����γ�(MgFe)H2������ĺϽ��γ���[31?32]����ʽ���㣺

��̬��ԭ�����������������㾧����������ͬ�����ƣ�Mg��Fe���嵥ԭ�������ļ���ֵ���ڱ�2����ʽ(3)��(4)����õ�MgH2��Mg10H20��(Mg9Fe)H20��(Mg4Fe2)H12��(Mg8Fe2)H20���������������ڱ�2�У��Ͻ��γ��ȵļ�������ͼ5����ͼ5�ɿ�����MgH2�ĺϽ��γ��ȵļ���ֵ(�C62.30 kJ/mol)�Դ���ʵ��ֵ((�C76.15��9.2) kJ/mol)[33]����������Ҫԭ�����ڱ��о��еļ���ֵΪ0 Kʱ��ֵ����ʵ��ֵ��Ϊ����670.7~707.9 K�IJⶨֵ����Fe�Ͻ�Fe������MgH2�У�MgH2��ϵ�ĸ��Ͻ��γ���ȴ���͡�

ͼ5 MgH2��(MgFe)H2�ĺϽ��γ���
Fig.5 Formation heat of MgH2 and (MgFe)H2 systems
���ڼ����Ԫ�Ͻ��⻯��ABnH2m�ĺϽ��γ��ȣ�MIEDEMA��[34]�����������Ľ��ƹ�ϵʽ��

������֮����Ϊ������������?H(ABn)Խ����ABnH2m���ȶ���Խ�͡�����Mg2FeH6���ԣ�ʵ���в�δ����Mg2Fe[5?7]����ˣ�����ʽ(5)���˼�����Ͻ��γ��ȣ����о��в�����ʽ���м��� [31?32]��
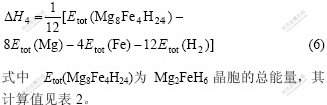
���㷢��Mg2FeH6�Ͻ��γ��ȵļ���ֵ(?124.87 kJ/mol)��ʵ��ֵ[24] (?79.2 kJ/mol)�����ƫ����SONG��[11]�ļ���ֵ(?124.15 kJ/mol)ʮ�� �ӽ���
Ϊ��������Mg2FeH6���MgH2��ϵ�ṹ�ȶ��Ե�Ӱ�죬���о��������ȶ��Բ�����Er�䣬��������ʾFe�Ͻ�MgH2��δ�Ͻ�MgH2�ĺϽ��γ��ȵı仯ֵ������㹫ʽΪ
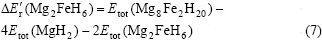
���ʽ(7)����Er��(Mg2FeH6)�ļ�����Ϊ45.358 7 kJ/mol��
������������ĺϽ��γ��ȣ����ֲ�ͬ����Fe������MgH2���γ� (MgFe)H2�����壬��δ����Fe�Ͻ�MgH2��ϵ��ȣ���ϵ?H�ľ���ֵ��С����ṹ�ȶ��Ա��[29?30]������Fe�Ͻ�MgH2��ϵ����������ߣ���Fe�Ͻ���ĥ��ĩ��������300 ��(573 K)�������½����ʵ�������[7]����Mg2FeH6��ĸ��Ͻ��γ���(?124.87 kJ/mol)����MgH2(?62.30 kJ/mol)����ṹ���ȶ��������µķ�����Mg2FeH6[5, 7]�����ʽ(7)�ļ���ֵ(45.3587 kJ/mol)Ϊ���������������ṹ���ȶ���Mg2FeH6��Ĵ��ڣ����MgH2��ϵ�Ľṹ�ȶ��Խ�һ�����͡�����Mg2FeH6��MgH2�������ϵ�У�MgH2��Ϊ������(��ʹFe������MgH2��)[7]��Mg2FeH6������Խ��٣�����������ȶ����ڵ�Mg2FeH6����������ã�ʹMgH2��ϵ�������ܽ�һ����ǿ����Ϻý����˱����������������ʵ��������Mg2FeH6��MgH2������ĩ�����з�ĩ������խ���¶ȷ�Χ���������ϴ���ʾ���Ľ�������ԼΪ0.0384%/��[7]��
2.6 ��ĥ������Fe�Ͻ���MgH2��ϵ���ܵ��ۻ���
��ĥMg��������϶�ȱ�ݣ�������H2��Mg������������������MgH2�������ܡ�̽�����ۻ��������о��м�������ࡢ��λȱ�ݱ���ı���Mgԭ��������H2ǰ�����̬(DOS)���ֲ�̬�ܶ�(PDOS)����ͼ6��ʾ����ͼ6(a)�ɼ���H2����ǰ�����Mg(0001)�������ƽ��ÿ��Mgԭ�ӵijɼ�����Ҫ�����ڷ����ܼ�EF��?7 eV�������䣬���У���?6~0 eV���䣬�ɼ�������Ҫ���Ը���Mg(s)��Mg(p)�Ĺ��ף�����?7~?6 eV����, �ɼ���������Ҫ���Ը���Mg(s)�Ĺ��ף�����������ȣ���λȱ�ݱ������ƽ��ÿ��Mgԭ�ӵijɼ���߶ȼ������ֲ����䲢���Ա仯(��ͼ6(b))��H2��������H2ֱ�Ӳ�������õ�Mgԭ��̬�ܶ����������ǰ�ķ����������仯(��ͼ6(c)��(d))������������λȱ�ݱ�����ԣ�����̬�ܶ���?7.5~?6.3 eV�ĵ��ܼ���������һ�³ɼ���(��6(c)��(d))������Ҫ����H(s)������Mg(s)�Ĺ��ף�Ԥʾ��H2�����Mgԭ�Ӽ���н����̶ȵ��������á�ͬʱ������������H2���ӵ�Ӱ�죬������λȱ�ݱ������Mg(s)�ķֲ�̬�ܶ���δ����H2��������ȣ���������ܼ��������ƶ�����Mg(p)�ķֲ�̬�ܶȱ仯��̫���ԡ���һ���Ƚ���H2ֱ�Ӳ�������õ�Mgԭ����H2���������̬�ܶ�ʱ����(��ͼ6(e))����ϵ̬�ܶȰ���ࡢ��λȱ�ݱ����˳��������ܼ������ƶ����ҷ����ܼ����µijɼ���߶�Ҳ����˳�������������������Mg(0001)�����H2������������ǿ����Ҫԭ��[35?36]��

ͼ6 H2��������ࡢȱ��Mg(0001)����ǰ����H2ֱ�Ӳ��������Mgԭ��ƽ��ÿ��ԭ�ӵ���̬�ܶȼ��ֲ�̬�ܶ�
Fig.6 Total and partial densities of states of clean and vacancy defective Mg (0001) surfaces during process of H2 adsorption: (a), (b) Densities of states between metal atom and Mg (0001) surface before H2 adsorption; (c), (d) Densities of states between metal atom and Mg (0001) surface after H2 adsorption; (e) Total density of states between metal atom and Mg (0001) surface after H2 adsorption; (f) Partial density of states between metal atom and Mg (0001) surface before H2 adsorption
�������������֣�H2������Mg(0001)����ǰ����H2ֱ�Ӳ�������õ�Mgԭ�ӵ�̬�ܶȱ仯��Ҫ������s����ķֲ�̬�ܶȱ仯������Ԥʾ�Ž���Mgԭ�ӵ�s������Ӷ�H2��������Ӱ��ϴ�Ϊ�ˣ���һ���Ƚ���Mg(0001)����δ����H2ʱ�������Mgԭ��s����ķֲ�̬�ܶȣ���ͼ6(f)��ʾ����?7~?4 eV�������䣬������Mg(s)�ķֲ�̬̬�ܶ����Ը��ڿ�λȱ�ݱ���ģ�����?4~0 eV���䣬����ȴ��Ȼ�෴���Կ�λȱ��Mg������ԣ����ܼ����϶�ijɼ�����������ܼ�EF��ת�ƣ�ʹ�����ܼ�������ɼ��ĵ������������࣬�����ܹ����Mg�ı������[37]����ܿ��ܾ��ǿ�λȱ�ݵĴ�����ǿ��Mg�����H2������������Ҫԭ��
Ϊ�о�Fe�Ͻ���MgH2��ϵ�������ܵ�ԭ���о��м�����Fe������MgH2�屶����ģ�͵���̬�ܶȼ���Ӧԭ�ӵķֲ�̬�ܶȡ��ڼ���ģ���У�Mg��H��Fe�ֱ�ռ�ݲ�ͬ�IJ��ȼ�ԭ��λ�ã�������ͼ2(b)��ʾ��Feδ����ʱ��MgH2�ڷ����ܼ�(EF)��?7.0 eV��������Χ�ڣ��ɼ����Ӵ��ڼ�����Ҫ�ijɼ���(��ͼ7(a))����0~?3.0 eV���䣬��ɼ�����ҪΪH(s)��Mg(p)�Լ�����Mg(s) �Ĺ��ף���?3.0 eV~ ?4.0 eV���䣬�ɼ�����Ҫ����H(s)��Mg(s)�Լ�����Mg(p) �Ĺ��ף�����?4.0 eV~?7.0 eV���䣬�ɼ�������Ҫ��H(s)��Mg(s)���Ľ������0~3.0 eV��������Χ�ڣ���1���ܿ�����϶�����������߷ֱ�Ϊ�ɼ���ͷ��ɼ��塣Fe������MgH2�У���̬�ܶȷ��������Եĸı�(��ͼ7(b)��7(c)��ʾ)����仯�У�1) ��϶�����ڷ����ܼ�(EF)����?2.0~?1.0 eV���䣬�������Ա�խ�� 2) ��?1.0~0.5 eV��Χ����Ҫ����ΪFe(d)�ĵ������ԣ���Fe������5.0%���ԣ�Mg2(p)��H3(s)�������Ĺ��ף���������10.0%ʱ����Mg2(p)��H3(s)���������⣬Mg3(p)��H1(s)��H2(s)Ҳ���������ף�3) Fe������Ϊ5.0%ʱ����?4.0~?6.0 eV���䣬H3�ijɼ���߶����ӣ�Fe������Ϊ10.0%ʱ����ͬһ���䣬H1��H3�ijɼ���߶Ⱦ����ӣ�4) EF������Mg(s)��Mg(p)��H(s)�ķֲ�̬�ܶȵĸ߶��������͡�����̬�ܶȵķ������������Fe������MgH2�У�Feԭ�������һ���ڵ�Hԭ��֮�������ǿ�ҵijɼ����ã���Mgԭ��������ڵ�Hԭ��֮��ijɼ�����������������

ͼ7 Mg10H20 (a)��(Mg9Fe)H20 (b)��(Mg8Fe2)H20 (c)����̬�ܶ���ֲ�̬�ܶ�
Fig.7 Total and partial densities of states of Mg10H20 (a), (Mg9Fe)H20 (b) and (Mg8Fe2)H20(c)
Ϊ��һ���о�Feԭ��������ڵ�Hԭ�ӵ�����ã���������Fe�ڽ���H1��H3ԭ���Լ���Mg���ڵ�H2ԭ��(��ͼ2(b))��˫���λ��ƽ����λ�γ��ܡ�Fe��MgH2�й�����Ϊ0.0%��5.0%��10.0%ʱ��H1��H2��H3ԭ��˫���λ��ƽ����λ�γ��ܦ�E(2H)�ֱ�����ʽ����[38]��
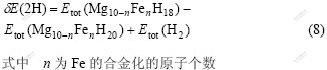
�磺Fe��MgH2�й�����Ϊ0.0%ʱ��n=0��������Ϊ5.0%ʱ��n=1, ������Ϊ10.0%ʱ��n=2��Etot(Mg10?nFenH18)Ϊ�γ�˫���λ��Mg10?nFenH18 �������������� Etot(Mg10?nFenH20)Ϊδ�γ�˫���λ��Mg10?nFenH20 ����������������������ͼ8��ʾ����ͼ8�ɼ���Fe�Ͻ�MgH2��ϵ����Fe�ڽ���H1��H3ԭ�ӵ�˫���λ��ƽ����λ�γ��ܱȶ�Ӧ��δ�Ͻ�ʱH1��H3�Ŀ�λ�γ��ܴ�������γ�H1��H3��λ���Ѷ����ӣ�Hԭ�ӽ����ͷţ���H2ԭ�ӵ�˫���λ��ƽ����λ�γ����ڽ���Fe�Ͻ�ȴС�ںϽ�ǰ�ģ�������γ�H2��λ���Ѷȼ�С��Fe�Ͻ�Hԭ�������ͷš�������Fe�Ͻ�Ũ�ȵ����ӣ���Fe�ڽ���H1��H3ԭ�ӣ�Hԭ���ͷŵ����׳̶������ı䣬H3ԭ�ӽ���
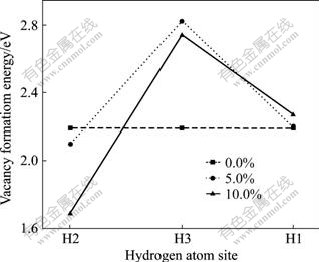
ͼ8 ��ͬλ��Hԭ�ӵĿ�λ�γ���
Fig.8 Vacancy formation energy of hydrogen atom in different kinds of sites
ͼ9��ʾΪFe������MgH2ǰ��ģ��(110)��ĵ����ܶȷֲ����Ƚ�ͼ9(a)��(b)��(c)�ɼ���Fe������MgH2�У�����1��Feԭ��ʱ��MgH2��Fe-H3֮����ڽ�ǿ������ã� Mg3-H2, Mg3-H1֮��������û�����Ա仯����Mg2-H2��Mg2-H3֮�������ü���������2��Feԭ��ʱ��MgH2��Fe-H3��Fe-H1֮������ڽ�ǿ������ã�Mg3-H2֮�������ñ仯�����ԣ���Mg2-H2��Mg2-H3, Mg3-H1֮�������ü�����
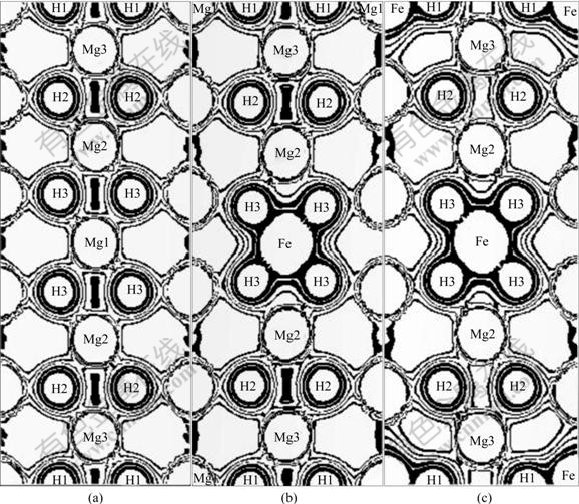
ͼ9 Mg10H20��(Mg9Fe)H20��(Mg8Fe2)H20��(110)��ĵ����ܶȷֲ�
Fig.9 Charge distributions on (110) plane of Mg10H20(a), (Mg9Fe)H20(b) and (Mg8Fe2)H20(c)
3 ����
1) H2�����Mg(0001)������ֽ��������������������Mg������H2����������ת�ƣ�Mg(0001)������ڿ�λȱ�ݣ���H2����������������ǿ������Mg������H2ת�Ƶĵ�������ࣻFe������MgH2���γ�(MgFe)H2������ͺϽ�ʱ�γ�����Mg2FeH6�࣬MgH2��ϵ�Ľṹ�ȶ��Ծ����ͣ���ϵ�������ܶ�Ӧ��ǿ��
2) ��λȱ������H2������Mg���棬��Mg(0001)�������ϲ���H2ֱ�Ӳ����������õĽ���ԭ���ڷ����ܼ�(EF)����s����ijɼ�������������ء�
3) MgH2��ϵ�У���Ͻ�Ԫ��Fe���ڵ�Hԭ���γɿ�λ���Ѷ����ӣ�Hԭ�ӽ����ͷţ�����Mg���ڵ�Hԭ���γɿ�λ���Ѷȼ�С��Hԭ�������ͷţ�Fe�Ͻ�MgH2��ϵ����������ǿ����Ҫԭ������Mg��H֮����ڽ����ijɼ����á�
REFERENCES
[1] ZALUSKA A, ZALUSKI L, STR?M-OLSEN J O. Nanocrystalline magnesium for hydrogen storage[J]. J Alloys Comp, 1999, 288(1/2): 217?225.
[2] JURCZYK M, SMARDZ L, OKONSKA I, JANKOWSKA E, NOWAK M, SMARDZ K. Nanoscale Mg-based materials for hydrogen storage[J]. Int J Hydrogen Energy, 2008, 33(1): 374?380.
[3] SHANG C X, BOUOUDINA M, SONG Y, GUO Z X. Mechanical alloying and electronic simulations of (MgH2+M) systems (M=Al, Ti, Fe, Ni, Cu and Nb) for hydrogen storage[J]. In J Hydrogenenergy, 2004, 29(1): 73?80.
[4] LIANG G, HUOT J, BOILY S, VAN NESTE A, SCHULZ R. Catalytic effect of transition metals on hydrogen sorption in nanocrystalline ball milled MgH2-Tm (Tm=Ti, V, Mn, Fe and Ni) systems[J]. J Alloys Comp, 1999, 292(1/2): 247?252.
[5] HUOT J, BOILY S, AKIBA E, SCHULZ R. Direct synthesis of Mg2FeH6 by mechanical alloying[J]. J Alloys Comp, 1998, 280(1/2): 306?309.
[6] HIGHTOWER A, FULTZ B, BOWMANN R C. Mechanical alloying of Fe and Mg[J]. J Alloys Comp, 1997, 252(1/2): 238?244.
[7] ZHOU D W, LI S L, VARIN R A, PENG P, LIU J S, YANG F. Mechanical alloying and electronic simulations of 2Mg-Fe mixture powers for hydrogen storage[J]. Mater Sci Eng A, 2006, 427(1/2): 306?315.
[8] SPRUNGER P T, PLUMMER E W. The interaction of hydrogen with simple metal surfaces[J]. Surf Sci, 1994, 307/309(1): 118?123.
[9] N?RSKOV J K, HOUM?LLER A, JOHANSSON P K, LUNDQUIST B I. Adsorption and dissociation of H2 on Mg surfaces[J]. Phys Rev Lett, 1981, 46(4): 257?260.
[10] VEGGE T. Locating the rate-limiting step for the interaction of hydrogen with Mg(0001) using density-functional theory calculations and rate theory[J]. Phys Rev B, 2004, 70(3): 035412?035418.
[11] SONG Y, GUO Z X, YANG R, Influence of selected alloying elements on the stability of magnesium dihydride for hydrogen storage applications: A first-principles investigation[J]. Phys Rev B, 2004, 69(1): 094205?094215.
[12] PERDEW J P, WANG Y. Accurate and simple analytic representation of the electron-gas correction energy[J]. Phys Rev B, 45(23): 13244-13249.
[13] DELLEY B. Analytic energy derivatives in the numerical local-density-functional approach[J]. J Chem Phys, 1991, 94 (11): 7245?7250.
[14] PACK J D, MONKHORST H J. Special points for brillouin-zone integrations��A reply[J]. Phys Rev B, 16(4): 1748?1749.
[15] PAYNE M C, TETER M P, ALLAN D C. Iterative minization techniques for Ab initio total energy calculations: molecular dynamics and cojugate gradients[J]. Rev Mod Phys, 1992, 64(1/2): 1045?1097.
[16] SEGALL M D, LINDAN P L D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Condens Matter, 2002, 14(11): 2717?2743.
[17] MARLO M, MILMAN V. Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals[J]. Phys Rev B, 2000, 62(4): 2899?2907.
[18] WHITE J A, BIRD D M. Implementation of gradient-corrected exchange-correlation potentials in car-parrinello total-energy calculations[J]. Phys Rev B, 1994, 50(1): 4954?4957.
[19] VANDERBILT D. Soft self-consistent pseudopotentitals in a generalized eigenvalue formalism[J]. Phys Rev B, 1990, 41(11): 7892?7895.
[20] FRANSCIS G P, PAYNE M C. Finite basis set corrections to total energy pseudopotential calcaulations[J]. J Phys: Condens Matter, 1990, 19(2): 4395?4404.
[21] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Phys Rev B, 1976, 13(1): 5188?5192.
[22] HAMMER B, HANSEN L B, NORKOV J K. Improved adsorption energetics withen density-functional theory using revised perdew-burke-ernzerhof functionals[J]. Phys Rev B, 1999, 59(11): 7413?7421.
[23] BORTZ M, BERTHEVILLE B, BOTTQER G, YVON K.  Structure of the high pressure phase ��-MgH2 by neutron powder diffraction[J]. J Alloys Comp, 1999, 287(1/2): L4?6.
Structure of the high pressure phase ��-MgH2 by neutron powder diffraction[J]. J Alloys Comp, 1999, 287(1/2): L4?6.
[24] DIDISHEIM J J, ZOLLIKER P, YVON K, FISCHER P, SCHEFER J, GUBELMANN M, WILLIAMS A F. Dimagnesium iron(II) hydride, Mg2FeH6, containing octahedral FeH64? anions[J]. Inorg Chem, 1984, 23(1/2): 1953.
[25] HUOT J, HAYAKAWA H, AKIBA E. Preparation of the hydrides Mg2FeH6 and Mg2CoH5 by mechanical alloying followed by sintering[J]. J Alloys Comp, 1997, 248(1/2): 164?167.
[26] SELVAM P, YVON K. Synthesis of Mg2FeH6, Mg2CoH5 and Mg2NiH4 by high-pressure sintering of the elements[J]. In J Hydrogenenergy, 1991, 16(1): 615?631.
[27] BIRD D M, CLARCKE L J, PAYNE M C, STICH I. Dissociation of H2 on Mg(0001)[J]. Chem Phys Lett, 1993, 212(5): 518?524.
[28] JENSEN P, BLAS? X, ORDEJ?N P. First principles study of gold adsorption and diffusion on graphite[J]. Surf Sci, 2004, 564(1): 173?178.
[29] ZHOU D W, PENG P, LIU J S. First-principles calculations of dehydrogenating properties of MgH2-V systems[J]. Science in China: Series E, 2006, 49(2): 129?136.
[30] �ܵ���, �� ƽ, ����ˮ. MgH2-Ti��ϵ�ĵ�һԭ������[J]. �й���ɫ����ѧ��, 2005, 15(9): 1403?1410.
ZHOU Dian-wu, PENG Ping, LIU Jin-shui. First-principles calculation of dehydrogenating properties of MgH2-Ti systems[J]. The Chinese Journal of Nonferrous Metals, 2005, 15(9): 1403?1410.
[31] MEDVEDEVA M I, GORNOSTYREV Y N, NOVIKOV D L, MRYASOV O N, FREEMAN A J. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl[J]. Acta Mater, 1998, 46(10): 3433?3442.
[32] SAHU B R. Electronic structure and bonding of ultralight LiMg[J]. Mater Sci Eng B, 1997, 49(1/2): 74?78.
[33] BOGDANOV?? B, BOHMHAMMEL K, CHRIST B, REISER A, SCHLICHITE K. Thermodynamic investigation of the magnesium-hydrogen system[J]. J Alloys Comp, 1999, 282(1/2): 84?92.
[34] MIEDEMA A R. The electronegativity parameter for transition metals: heat of formation and charge transfer in alloys[J]. J Less-Common Met, 1973, 32(1/2): 117?152.
[35] IMAI Y, MUKAIDA M, TSUNODA T. Comparison of density of states of transition metal disilicides and their related compounds systematically calculated by a first-principle pseudopotential method using plane-wave basis[J]. Intermetallics, 2000, 8(4): 381?390.
[36] VAKHNEY A G, YARESKO A N, ANTONOV V N, NEMOSHKALENKO V V. The effect hydrogen on the electronic structure and cohesive properties of iron-based alloys doped by chromium and nickel[J]. Int J Hydrogen Energy, 2001, 26(5): 453?456.
[37] EALET B, GONIAKOWSKI J, FINOCCHI F. Water dissociation on a defective Mg(100) surface: Role of divacancies. Phys Rev B, 2004, 69(19): 195413?195421.
[38] KELLOU A, FERAOUN H I, GROSDIDIER T, CODDET C, AOURAG H. Energetics and electronic properties of vacancies, anti-sites, and atomic defects(B, C, and N ) in B2-FeAl alloys[J]. Acta Mater, 2004, 52(11): 3263?3271.
������Ŀ��������Ȼ��ѧ����������Ŀ(50771044)�������ص�����о���չ�ƻ�������Ŀ(2006CB605104)
�ո����ڣ�2008-04-23�������ڣ�2008-07-08
ͨѶ���ߣ��ܵ��䣬�����ڣ���ʿ���绰��13017297124��E-mail: zdwe_mail@yahoo.com.cn
(�༭ ��ѧ��)

