
J. Cent. South Univ. (2018) 25: 772-782
DOI: https://doi.org/10.1007/s11771-018-3782-z
Density functional investigation on structural and electronic properties of small bimetallic PbnAgn (n=2–12) clusters
LI Gao-feng(李高锋)1, 2, 3, WANG Jia-ju(王家驹)1, 2, 3, CHEN Xiu-min(陈秀敏)1, 2, 3,
YANG Hong-wei(杨红卫)1, 2, 3, YANG Bin(杨斌)1, 2, 3, XU Bao-qiang(徐宝强)1, 2, 3, LIU Da-chun(刘大春)1, 2, 3
1. State Key Laboratory of Complex Nonferrous Metal Resources Clear Utilization,Kunming University of Science and Technology, Kunming 650093, China;
2. National Engineering Laboratory for Vacuum Metallurgy, Kunming University of Science andTechnology, Kunming 650093, China;
3. Yunnan Provincial Key Laboratory for Nonferrous Vacuum Metallurgy, Kunming University of Science and Technology, Kunming 650093, China
 Central South University Press and Springer-Verlag GmbH Germany, part of Springer Nature 2018
Central South University Press and Springer-Verlag GmbH Germany, part of Springer Nature 2018
Abstract: Structural and electronic properties of PbnAgn (n=2–12) clusters were investigated by density functional theory with generalized gradient approximation at BLYP level in DMol3 program package. The optimized bimetallic PbnAgn (n=2–12) clusters were viewed as the initial structures, then, those were calculated by ab initio molecular dynamics (AIMD) to search possible global minimum energy structures of PbnAgn clusters, finally, the ground state structures of PbnAgn (n=2–12) clusters were achieved. According to the structural evolution of lowest energy structures, Ag atoms prefer gather in the central sites while Pb atoms prefer external positions in PbnAgn (n=2–12) clusters, which is in excellent agreement with experimental results from literature and the application in metallurgy. The average binding energies, HOMO-LUMO gaps, vertical ionization potentials, vertical electron affinities, chemical hardness η, HOMO orbits, LUMO orbits and density of states of PbnAgn (n=2–12) clusters were calculated. The results indicate that the values of HOMO-LUMO gaps, vertical ionization potentials, vertical electron affinities and chemical hardness η show obvious odd-even oscillations when n≤5, PbnAgn (n=2–12) clusters become less chemically stable and show insulator-to-metal transition with the variation of cluster size n, PbnAgn (n≥9) cluster are good candidates to study the properties of PbAg alloys. Those can be well explained by the density of states (DOS) distributions of Pb atoms and Ag atoms between –0.5 Ha and 0.25 Ha in PbnAgn (n=2–12) clusters.
Key words: density functional theory; PbnAg n (n=2–12) clusters; ab initio molecular dynamics; ground state structure
Cite this article as: LI Gao-feng, WANG Jia-ju, CHEN Xiu-min, YANG Hong-wei, YANG Bin, XU Bao-qiang, LIU Da-chun. Density functional investigation on structural and electronic properties of small bimetallic PbnAgn (n=2–12) clusters [J]. Journal of Central South University, 2018, 25(4): 772–782. DOI: https://doi.org/10.1007/s11771-018-3782-z.
1 Introduction
Cluster is composed of two or more than two atoms, molecules and ions, and its properties varied from different sizes. Conventionally, physical and chemical methods are widely used to fabricate clusters. Recently, clusters arise much attention from scientists and scholars due to its unique properties in nanotechnology. Comparing with the traditional materials, clusters have many popular properties, such as used as adsorbent [1–9], catalysis [10], nanoscale materials in building blocks [11] and optical properties [12].
Many researchers have done much investigations on pure Pb clusters, and obtained a great deal of achievements on experiments and theory. TCHAPLYGUINE et al [13] used the synchrotron-based X-ray photoelectron spectroscopy to study the electronic structures and structural evolutions of neutral Pb clusters, and the results show that neutral Pb clusters have an insulator-to-metal transition as the cluster size n increases. LI et al [14] have studied the fragmentation behavior and ionization potentials of Pbn (n≤20) clusters, using ab initio calculation to investigate the binding energies, second differences in energy, and HOMO-LUMO gaps, especially fragmentation energies and ionization potentials. The results show that the fragmentation channels of large clusters and the calculated ionization potentials are in agreement with the experimental data. GINGERICH et al [15] investigated thermodynamics of Pb2, Pb3, and Pb4 clusters by mass spectrometry. Structures, stabilities and electronic properties of FePbn (n=1–14) clusters were theoretically studied by BAI et al [16], the results indicate that the Fe atom prefers implant in the interior of the clusters as the number of Pb atom increases.
Silver belongs to coinage metals with many unique properties. The Agn clusters have attracted many researchers’ attentions [17–20]. LI et al [21] investigated the optical responses of Ag7 cluster, Cu7 cluster and bimetallic Ag–Cu clusters with single excitation and multiple excitation generation (MEG) by SAC-CI, the results are in excellent agreement with experimental spectra. MA et al [22] employed DFT to study the optical and electronic properties of Ag13 cluster, Ag12Cu cluster and Cu13 cluster. TAFOUGHALT et al [23] investigated the structural and electronic properties of bimetallic AgnCum (m+n≤8) clusters, and found that s-d hybridization plays an vital role in silver–gold clusters and the highest occupied molecular orbital is affected by s and d electrons by increasing the concentration in the bimetallic clusters.
So far, many pure Pbn clusters, Agn clusters and the pure clusters doped a impurity atom have been widely investigated by their structural and electronic properties, but these studies were limited to a small number of their candidate structures because determining the lowest energy structures of clusters is an essential step in understanding their properties. Empirical method and semi-empirical method [24] were widely used to generate isomers of clusters. Using ab initio molecular dynamics is a good way to search the global energy minimum structure of cluster [25, 26]. Currently, Ag, Cu, Sn, Zn, As, Sb and Bi elements are the common impurities in crude lead [27]. Furthermore, the refining of crude lead has many disadvantages in fire refining process and electrolytic process [28], for example, high metal content in tailing, low direct rate of crude lead and environmental pollution in fire refining process, high energy consumption and long production duration in electrolytic process. But vacuum metallurgy has many advantages [28], such as short flow, environmental protection and low cost. In addition, to the best of our knowledge, scare works reported the binary PbnAgn (n=2–12) clusters. However, it is very important and urgent to systematically study the PbnAgn clusters because this may provide valuable information for separation of Pb–Ag alloys using vacuum metallurgy and synthesis of Pb–Ag materials.
2 Computational details
In this work, all calculations were implemented in spin-polarized DFT in the DMol3 program package [29] of Material Studio software. Using generalized gradient approximation (GGA) [30–33] together with Beke’s gradient-corrected exchange potential and Lee-Yang-Parr gradient- corrected exchange potential (BLYP) [29] to treat the exchange and correlation potential, the effective core potential considering DFT semi-core potential (DSSP) [33] and double numerical basis with d-polarization function (DND) [29] were adopted. Self-consistent field calculations were finished with the SCF tolerance standard of 10–6 Ha on the total energy. The density mixing standards for charge and spin are 0.2 and 0.5, respectively, the smearing is 0.005 Ha. In the geometry optimization and energy calculations, the energy tolerance is 10–5 Ha per atom, the maximum force is 0.002 Ha/ , the maximum displacement is 0.005 .
, the maximum displacement is 0.005 .
In order to search the structures with possible global minimum energy for each bimetallic PbnAgn (n=2–12) clusters, the ab initio molecular dynamics simulations were used. Dynamic parameters were set, ensemble is NVT, temperature is 300 K, total simulation time is 10 ps with 1 fs for each step and the thermostat is Massive GGM. It is indispensable to mention that the stable structures were checked out without imaginary frequency.
In order to verify that the scheme for bimetallic PbnAgn (n=2–12) clusters is reliable, the bond lengths, binding energies, frequencies and vertical ionization potentials of Ag2 and Pb2 dimers were calculated. The computational results and reported results are given in Table 1. It is interesting to note that the computational results in this work are in good agreement with reported results from literature except for the underestimated value of Eb for Ag2 dimer and Pb2 dimer. Therefore, our computational method is reliable and accurate enough for the investigation of bimetallic PbnAgn (n=2–12) clusters.
3 Results and discussion
3.1 Lowest-energy structures
In order to obtain the ground state structures of PbnAgn (n=2–12) clusters, we optimized the local lowest energy isomers of PbnAgn clusters after ab initio molecular dynamics, then the energies of isomers for each size were calculated and compared, finally, the lowest-energy structures were obtained. The lowest-energy structures of PbnAgn (n=2–12) clusters are shown in Figure 1.
Table 1 Bond lengths (R), binding energies (Eb), frequencies (w) and vertical ionization potentials (IP) of Ag2 and Pb2 dimers from our DFT calculations at GGA/BLYP level and that of reported results from literature

As we can see from Figure 1, Pb2Ag2 cluster (C2V) is in the shape of quadrihedron, Ag atoms are adjacent to Ag atoms while the same to Pb atoms. Pb3Ag3 cluster is in the shape of bipyramid in which two of Pb atoms are located on the apexes. Obviously, Pb3Ag3 cluster (C2V) is originated from Pb2Ag2 cluster by adding one Pb atom and one Ag atom. Pb4Ag4 cluster (C2d) is a two layers structure at first glance, the Ag atoms occupy peripheral sites.
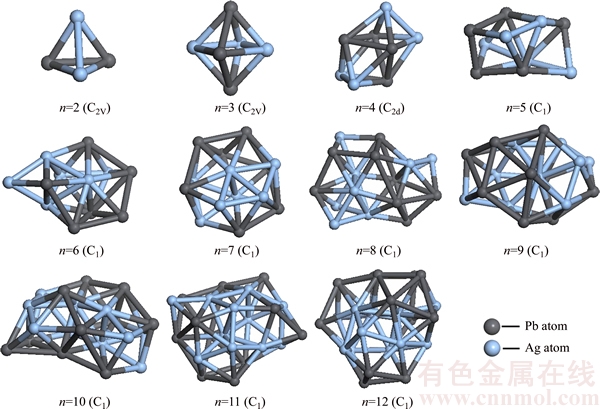
Figure 1 Optimized lowest-energy structures of PbnAgn (n=2–12) clusters
Pb5Ag5 cluster (C1) is also a distorted two layer structure with two capped quadrangles. Pb6Ag6 cluster (C1) is shaped like a prolate structure, Ag atoms have the tendency of gathering together. Pb7Ag7 cluster (C1) consists of bi-layer stacked hexagonal structures which two Ag atoms are in the centre of the cluster. Pb8Ag8 cluster (C1) is a prolate structure, it is originated from Pb6Ag6 cluster by capped two Ag atoms and two Pb atoms. Pb9Ag9 cluster (C1) is made up of ellipsoid structure. Pb10Ag10 cluster (C1) likes a hollow disk with two layers. Pb11Ag11 cluster (C1) is a cylindrical structure with Pb atoms been separated by Ag atoms. Pb12Ag12 cluster (C1) is a close-packed structure and all Ag atoms gather together in the center. Overall, there are a common tendency that Ag atoms tend to gather together and fall into the centre of the PbnAgn (n=2–12) clusters based on the structural evolution.
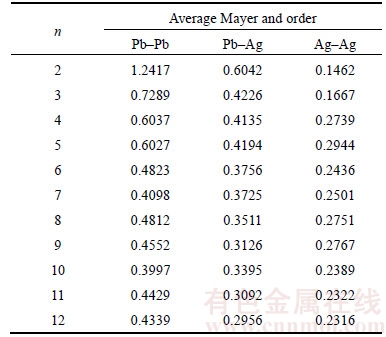
The concept of Mayer bond orders was put forward by Mayer in 1986. It is a good index to describe the relative stability of chemical bond in molecule. Average Mayer bond orders of PbnAgn (n=2–12) clusters are given in Table 2. It is interesting to note that Mayer bond orders of Pb—Pb bonds are greater than that of Pb—Ag bonds, and bond orders of Ag—Ag bonds are the smallest among them for each cluster. But Mayer bond orders of Pb—Pb bonds and Pb—Ag bonds decrease versus the cluster size n of PbnAgn (n=2–12) clusters, while the Ag—Ag bonds show the opposite trend, it plays a crucial role in the structural evolution of PbnAgn (n=2–12) clusters with Ag atoms tend to gather together and fall into the centre of the PbnAgn (n=2–12) clusters. Furthermore, all the bond orders become weaker as the cluster size n of the PbnAgn (n=2–12) clusters increases, the Mayer bond orders of Pb—Pb bonds, Pb—Ag bonds and Ag—Ag bonds tend to be stable when n≥10. Only in this way, could the PbnAgn (n=2–12) clusters keep the most stable structures.
Table 2 Average Mayer bond orders of PbnAgn (n=2–12) clusters
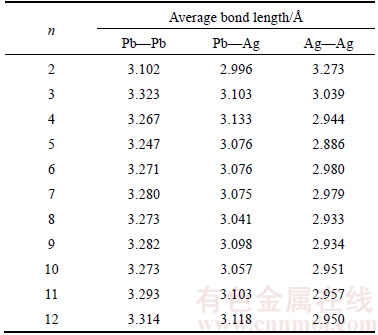
Average bond lengths of PbnAgn (n=2–12) clusters are shown in Table 3. The average bond lengths of Pb—Pb bonds are greater than that of Pb—Ag bonds, and the average bond lengths of Pb—Ag bonds are greater than that of Ag—Ag bonds for PbnAgn (n=2–12) clusters. With the variation of size n in PbnAgn (n=2–12) clusters, the bond lengths of Pb—Pb bonds increase, and the bond lengths of Ag—Ag bonds decrease, the bond lengths of Pb—Ag bonds maintain stable. With the variation of size n in PbnAgn (n=2–12) clusters, the Ag—Ag bonds are easier to bond compared with that of Pb—Pb bonds and Ag—Pb bonds. It is also in accord with the structural evolution of PbnAgn (n=2–12) clusters discussed above. The bond lengths of Pb—Pb bonds, Pb—Ag bonds and Ag—Ag bonds tend to be stable when n≥10. The trend of average bond lengths is similar to Mayer bond orders in PbnAgn (n=2–12) clusters. It would be the reasons for Ag atoms tend to gather together and fall into the centre of the PbnAgn clusters as the size n increasing.
Table 3 Average bond lengths of PbnAgn (n=2–12) clusters
In a previous work, PARGA et al [36] reported the experimental investigation of recovering refractory Ag and Au with recycling lead. The flowsheet is shown in Figure 2. The lead acts as a solvent during the smelting process. The reaction mechanism can be briefly described as follows [36]:
2FeS2+15PbO=Fe2O3+4SO2+15Pb;
FeS2+5PbO=FeO+2SO2+5Pb;
PbO+Fe=Pb+FeO;
4SO3+4Na2CO3=4CO2+4Na2SO4;
7PbO+FeS2+Na2CO3=7Pb+2Na2SO4+FeO+2CO2;
6PbO+2FeS+2SiO2=6Pb+2FeSiO3+2SO2;
Ag2S+Na2CO3+C=2Ag+Na2S+CO2+CO;
Ag2S+2PbO=2(Pb, Ag)+SO2.
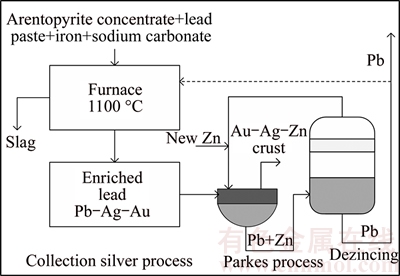
Figure 2 Flowsheet of recovering precious metals with recycling lead [36]
The reactions above indicate that the products of smelting process can be divided into gas phase, slag phase and lead bullion phase. Then, the lead bullion is obtained by separating from slag, bullion is consisted of large proportion of silver and little proportion of gold. A crust was formed, in which Ag and Au were extracted into lead phase. In other words, the use of lead acts as concentrating agent that Ag and Au are surrounded by lead. However, our calculated structural evolution of PbnAgn (n=2–12) clusters is in excellent agreement with the experimental results. The structural evolution of PbnAgn (n=2–12) clusters would provide much guidance for low-cost metals enrich precious metals during metallurgical process and synthesis of bimetallic alloys with controllable microstructures.
3.2 Electronic properties of PbnAgn (n=2–12) clusters
The binding energy Eb per atom of the lowest-energy structures of PbnAgn (n=2–12) clusters is given in Figure 3. The average binding energy (Eb) is defined as [37, 16]:
Eb=[nE(Pb)+nE(Ag)–E(PbnAgn)]/(2n)
where E(Pb), E(Ag) and E(PbnAgn) are the total energies of Pb atom, Ag atom and PbnAgn cluster, respectively. The binding energy is a measurement of total thermodynamics stability for clusters. From Figure 3, the average binding energies of PbnAgn (n=2–12) clusters increase as a function of the cluster size n. The trend of Eb indicates that the stability of PbnAgn clusters increase with the size n of the clusters. The phenomenon can be expressed as the atoms have a greater interaction per atom, because the nearest neighbors increase with the growing size n [38]. There is an exception that the average binding energy of Pb4Ag4 cluster is almost equal to that of Pb5Ag5 cluster, it may be originated from that Pb4Ag4 cluster is more compact than Pb3Cu3 cluster, and the compactness of Pb4Ag4 is almost equal to that of Pb5Ag5 cluster. The clusters tend to be stable when n≥9. It is suggested that the Pb9Ag9 (n≥9) clusters are representative candidates if we want to study the metallurgical behavior and other properties of PbAg alloys.
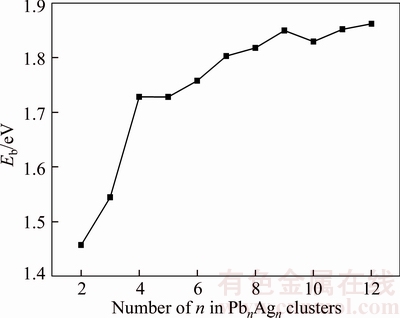
Figure 3 Size dependence of average binding energies for PbnAgn (n=2–12) clusters
The energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are depicted in Figure 4. The HOMO-LUMO gap shows the chemical stability of a cluster, a large gap means the cluster is more stable and abundant because a large energy is needed if an electron excites from valence band to conduction band. A cluster with a large HOMO-LUMO gap is usually equipped with closure of the electronic shell and regarded as the magic cluster. As we know, the electron configurations of Pb atom and Ag atom are 6s26p2 and 4d105s1, respectively. In Figure 4, it implies odd-even alternations, the PbnAgn cluster with even-numbered cluster size n has an even number of valence electrons and has a larger value of HOMO-LUMO gap than the neighboring odd ones except for Pb6Ag6 cluster. The effect can be explained by electron pairing [23]. The HOMO- LUMO gaps of Pb4Ag4 cluster, Pb8Ag8 cluster and Pb8Ag8 cluster are obviously higher than that of their neighbors. Indeed, Pb4Ag4 cluster, Pb8Ag8 cluster and Pb10Ag10 cluster with 20, 40 and 50 valence electrons are in accord with the major shell closing of electron shell model. Overall, the HOMO-LUMO gaps obviously decrease with the increasing cluster size n, it suggests a transition from non-metallic behavior to metallic one and become less chemically stable. This trend of nonmetal-to-metal transition is similar with the result of pure lead clusters with the theoretical calculation by WANG et al [29] and experimental study by SENZ et al [39].

Figure 4 Size dependence of HOMO-LUMO gaps for PbnAgn (n=2–12) clusters
The vertical ionization potentials (VIP) and vertical electron affinities (VEA) as a function of cluster size for PbnAgn (n=2–12) clusters are plotted in Figures 5 and 6, respectively. The vertical ionization potential and vertical electron affinity can be defined as [37]
EVIP=E(PbnAgn+)–E(PbnAgn);

Figure 5 Size dependence of vertical ionization potentials for PbnAgn (n=2–12) clusters
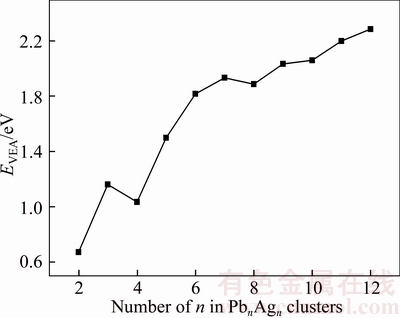
Figure 6 Size dependence of vertical electron affinities for PbnAgn (n=2–12) clusters
EVEA=E(PbnAgn)–E(PbnAgn-)
where EVIP is the vertical ionization potential, EVEA is the vertical electron affinity, E(PbnAgn+), E(PbnAgn) and E(PbnAgn-) are the total energy of PbnAgn+ cluster, PbnAgn cluster and PbnAgn- cluster, respectively.
Vertical ionization potential (VIP) is also to describe the chemical stability of small clusters, and higher vertical ionization potential corresponds to a deeper energy level of highest occupied molecular orbit. It means that neutral cluster loses an electron and becomes a positive cluster needing more energy during the ionization process. A higher vertical electron affinity (VEA) indicates that the more energy could be released if neutral cluster gets an electron from others, and the corresponding anion cluster could be more difficult to occur. As we can see from Figures 5 and 6, the clusters with even numbered n are bigger than their neighbors with odd numbered n for vertical ionization potentials when n≤5, while the vertical electron affinities of corresponding clusters show the opposite trend. Overall, vertical ionization potentials tend to increase with the variation of cluster n in PbnAgn (n=2–12) clusters, while vertical electron affinities show the opposite trend.
According to the molecular orbital theory, the Koopmans approximation and the finite difference approximation, chemical hardness η can be approximated as [37]
η=(EVIP–EVEA)/2
where the EVIP and EVEA are vertical ionization potential and vertical electron affinity of the cluster, respectively. The chemical hardness for lowest energy structures of PbnAgn (n=2–12) clusters are calculated and depicted in Figure 7. From Figure 7, we can see that the chemical hardness show an odd-even oscillation when n≤5. Overall, the chemical hardness decreases with the increasing cluster size n, showing that the chemical stability decrease with increasing cluster size n. It is in agreement with the trend of HOMO-LUMO gaps.
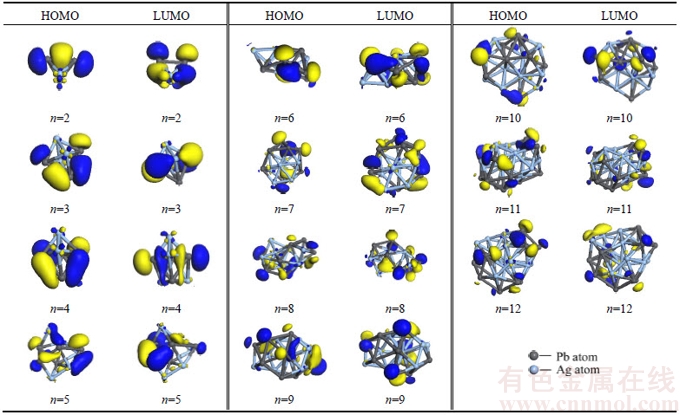
In order to get well understanding of natural chemical bonding in PbnAgn (n=2–12) clusters, the spatial orientations of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are depicted in Table 4. As we can see, the electron clouds mainly distribute among Pb atoms. The distribution of electron clouds among peripheral atoms are much and well-proportioned when n≤9 while a few among internal atoms. There are a few electron clouds among peripheral atoms discretely when 10≤n≤12, almost no electron cloud among internal atoms. The distribution of electron clouds among HOMO and LUMO could be used to explain why the PbnAgn (n=2–12) clusters become less and less chemical stable with the variation of cluster n increased. It is also in excellent accord with the trend of HOMO-LUMO gaps and chemical hardness η. The scare electron clouds among Ag atoms could be used to explain why VIPs, VEAs, chemical hardness η of PbnAgn (n=2–12) clusters do not show obvious odd-even oscillations with the odd-numbered valence electrons of Ag atom and even-numbered valence electrons of Pb atom in PbnAgn (n=2–12) clusters, it may be originated from that the valence electrons of Ag atoms make little contributions to the chemical stability of PbnAgn (n=2–12) clusters while the Pb atoms make huge contributions to it.
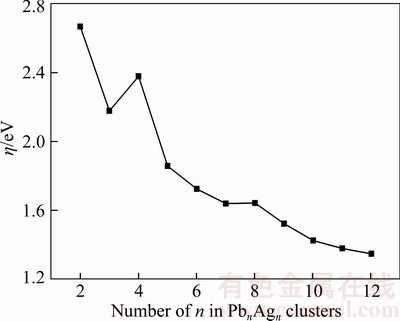
Figure 7 Size dependence of chemical hardness for PbnAgn (n=2–12) clusters
Table 4 Spatial orientations of HOMOs and LUMOs for PbnAgn (n=2–12) clusters
To further understand the electronic properties of PbnAgn (n=2–12) clusters, the density of states (DOS) were plotted in Figure 8. The density of states can indicate detail information of valence electron distribution of PbnAgn (n=2–12) clusters. Within the energy level from –3.5 Ha and –0.4 Ha, Ag atoms only have two strong localized s and p peaks, Pb atoms only have one strong localized d peak and have little contributions from other electrons, also those electrons do not have any changes with the variation of cluster size n in PbnAgn (n=2–12) clusters, this indicates that the electrons within the interval can hardly changed and is not important in the structural evolution and electronic properties of PbnAgn (n=2–12) clusters. Therefore, the electrons between –3.5 Ha and –0.4 Ha should be ignored. However, we should put much attention on the electrons between –0.5 Ha and 0.25 Ha. From the Figure 8, the 5s orbitals of Ag atoms and 6p orbitals of Pb atoms are overlapped between –0.075 Ha and –0.025 Ha, and the 4p orbitals of Ag atoms and 5d orbitals of Pb atoms are overlapped between 0.2 Ha and 0.3 Ha, indicating a strong hybridization in Pb2Ag2 cluster. For Pb3Ag3 cluster, the superposition of 4p orbitals of Ag atoms and 6s orbitals of Pb atoms between –3.5 Ha and –2.5 Ha, 5s orbitals of Ag atoms and 6p orbitals of Pb atoms between –1.0 Ha and –0.05 Ha, and 4p orbitals of Ag atoms and 5d orbitals of Pb atoms between 1.25 Ha and 2.25 Ha refer to strong hybridizations. Similarly, the hybridizations of 5s orbital of Ag atoms and 6p orbital of Pb atoms between –0.085 Ha and 0.025 Ha, and 5s orbitals of Ag atoms and 6p orbitals of Pb atoms between 0.05 Ha and 0.15 Ha for Pb4Ag4 cluster, 5s orbitals of Ag atoms and 6p orbitals of Pb atoms between –0.115 Ha and –0.25 Ha for Pb5Ag5 cluster. For the PbnAgn (n=6–12) clusters, there are no hybridization or very weak hybridizations and the density of states (DOS) are mainly from the valence electrons of Pb atoms except for the localized d orbitals of Ag atoms between –0.4 Ha and 0.3 Ha, those could well explain why the clusters (n=2–5) are more chemical stable than other clusters (n=6–12) and odd-even oscillations due to the contribution of valence electrons of Ag atoms. In addition, when n≤4, the density of states (DOS) are very localized, when 5≤n≤12, the density of states (DOS) become continuous, which indicate that the metallicity of PbnAgn clusters become more and more strong and are in excellent agreement with the trend of HOMO-LUMO gaps and chemical hardness.
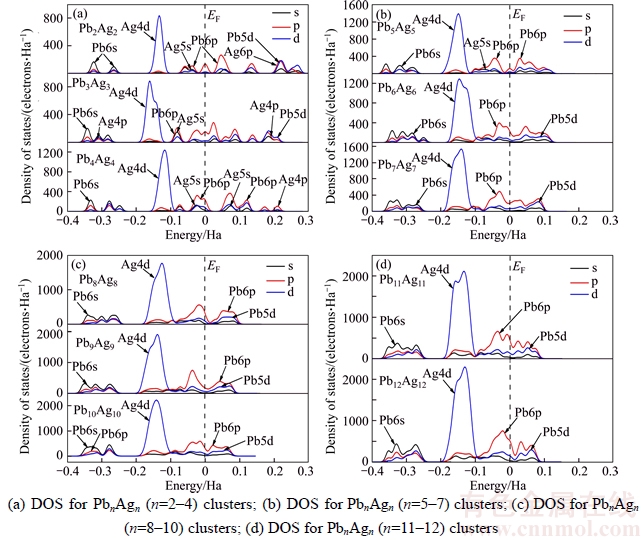
Figure 8 Size dependence of DOS for PbnAgn (n=2–12) clusters:
4 Conclusions
In this work, all the calculations were carried out by density functional theory (DFT) in DMoL3 package. The results can be summarized as follows:
1) With the lowest energy structural evolution of PbnAgn (n=2–12) clusters, the Ag atoms show the trend of falling into the centre of PbnAgn clusters and gathering together as the cluster size n increased. The bond lengths Pb—Pb bonds are greater than that of Pb—Ag bonds, while bond lengths Ag—Ag bonds are the shortest among all the bonds in PbnAgn (n=2–12) clusters. The variation of bond lengths is consistent with the change of Mayer bond orders. These contribute to the structural evolution that Ag atoms are surrounded by Pb atoms. With the mechanism, Pb is an excellent solvent to enrich previous metals.
2) The average binding energies increase with the increasing cluster size n when n≤8. The PbnAgn (n≥9) clusters are good candidates which could well perform physical and chemical properties of large PbAg alloys, and could be used to calculate the molecular interaction energy and the molecular interaction parameter of PbAg alloys under vacuum.
3) The HOMO-LUMO gaps, VIPs, VEAs and chemical hardness η of PbnAgn clusters show an odd-even oscillation when n≤5, while do not show an odd-even oscillation when 6≤n≤12. There is a trend that the PbnAgn (n=2–12) clusters become less chemically stable. The density of states (DOS) distribution of Pb atoms and Ag atoms between –0.5 Ha and 0.25 Ha play a vital role in PbnAgn (n=2–12) clusters. Moreover, the nonmetal-to-metal transition of PbnAgn (n=2–12) clusters could provide important information for the synthesis of PbAg nanomaterials.
References
[1] SAMANTA P N, DAS K K. Adsorption of CO on pure and mixed clusters of tin and germanium up to five atoms: A theoretical study [J]. Computational and Theoretical Chemistry, 2012, 1000: 42–51.
[2] CORT S-ARRIAGADA D, OYARZ
S-ARRIAGADA D, OYARZ N M P, SANHUEZA L, TORO-LABBE A. Binding of trivalent arsenic onto the tetrahedral Au20 and Au19Pt clusters: Implications in adsorption and sensing [J]. The Journal of Physical Chemistry A, 2015, 119(26): 6909–6918.
N M P, SANHUEZA L, TORO-LABBE A. Binding of trivalent arsenic onto the tetrahedral Au20 and Au19Pt clusters: Implications in adsorption and sensing [J]. The Journal of Physical Chemistry A, 2015, 119(26): 6909–6918.
[3] de HAECK J, VELDEMAN N, CLAES P, JANSSENS E, ANDERSSON M, LIEVENS P. Carbon monoxide adsorption on silver doped gold clusters [J]. The Journal of Physical Chemistry A, 2011, 115(11): 2103–2109.
[4] ZHAO Shuang, LI Guo-zhi, LIU Jun-Na, REN Yun-lai, LU Wei-wei, WANG Ji. Density functional study of ethylene adsorption on small gold, palladium and gold-palladium binary clusters [J]. The European Physical Journal D, 2014, 68(9): 1–8.
[5] ZHAO Shuang, TIAN Xin-zhe, LIU Jun-na, REN Yun-lai, REN Yun-li, WANG Jian-ji. Density functional study of molecular hydrogen adsorption on small gold–copper binary clusters [J]. Journal of Cluster Science, 2015, 26(2): 491–503.
[6] SAMANTA P N, DAS K K. First principle study of the sensitivity of CO adsorption on pure and binary clusters of lead and silicon [J]. The European Physical Journal D, 2014, 68(7): 1–10.
[7] CORTS-ARRIAGADA D, TORO-LABB A. Insights into the use of Au19Cu and Au19Pd clusters for adsorption of trivalent arsenic [J]. Theoretical Chemistry Accounts, 2016, 135(3): 1–13.
[8] MONDAL K, MANNA D, GHANTY T K, BANERJEE A. Significant modulation of CO adsorption on bimetallic Au19Li cluster [J]. Chemical Physics, 2014, 428: 75–81.
[9] KUANG Xiang-jun, WANG Xing-qiang, LIU Gao-bin. The adsorptions of silver-doped small gold clusters toward carbon monoxide molecule [J]. Structural Chemistry, 2012, 23(3): 671–679.
[10] CHANG Chun-ran, WANG Yang-gang, LI Jun. Theoretical investigations of the catalytic role of water in propene epoxidation on gold nanoclusters: A hydroperoxyl-mediated pathway [J]. Nano Research, 2011, 4(1): 131–142.
[11] ALIVISATOS A P, JOHNSSON K P, PENG X, WILSON T E, LOWETH C L, BRUCHEZ M P J, SCHULTZ P G. Organization of 'nanocrystal molecules' using DNA [J]. Nature, 1996, 382(6592): 609–611.
[12] OLDENBURG S J, JACKSON J B, WESTCOTT S L, HALAS N J. Infrared extinction properties of gold nanoshells [J]. Applied Physics Letters, 1999, 75(19): 2897–2899.
[13] TCHAPLYGUINE M,  HRWALL G, ANDERSSON T, SVENSSON S, BJRNEHOLMB O, HUTTULA M, MIKKEL
HRWALL G, ANDERSSON T, SVENSSON S, BJRNEHOLMB O, HUTTULA M, MIKKEL M, URPELAINEN S, OSMEKHIN S, CAL
M, URPELAINEN S, OSMEKHIN S, CAL A, AKSELA S, AKSELA H. Size-dependent evolution of electronic structure in neutral Pb clusters—As seen by synchrotron-based X-ray photoelectron spectroscopy [J]. Journal of Electron Spectroscopy and Related Phenomena, 2014, 195: 55–61.
A, AKSELA S, AKSELA H. Size-dependent evolution of electronic structure in neutral Pb clusters—As seen by synchrotron-based X-ray photoelectron spectroscopy [J]. Journal of Electron Spectroscopy and Related Phenomena, 2014, 195: 55–61.
[14] LI Xiao-ping, LU Wen-cai, ZANG Qing-jun, CHEN Guang-ju, WANG C Z, HO K M. Structures and stabilities of Pbn (n≤20) clusters [J]. The Journal of Physical Chemistry A, 2009, 113(22): 6217–6221.
[15] GINGERICH K A, COCKE D L, MILLER F. Thermodynamic investigation of the lead molecules Pb2, Pb3, and Pb4 by mass spectrometry [J]. The Journal of Chemical Physics, 1976, 64(10): 4027–4033.
[16] BAI Yu-jie, CHENG Hai-ying, SUN Hou-qian, XU Ning, DENG Kai-ming. Structures, stabilities and electronic properties of FePbn (n=1–14) clusters: Density-functional theory investigations [J]. Physica B: Condensed Matter, 2011, 406(20): 3781–3787.
[17] YABANA K, BERTSCH G F. Optical response of small silver clusters [J]. Physical Review A, 1999, 60(5): 3809.
[18] KREIBIG U. Electronic properties of small silver particles: The optical constants and their temperature dependence [J]. Journal of Physics F: Metal Physics, 1974, 4(7): 999.
[19] HARB M, RABILLOUD F, SIMON D, RYDLO A, LECOULTRE S, CONUS F, RODRIGUES V,  C. Optical absorption of small silver clusters: Agn (n=4–22) [J]. The Journal of Chemical Physics, 2008, 129: 194108.
C. Optical absorption of small silver clusters: Agn (n=4–22) [J]. The Journal of Chemical Physics, 2008, 129: 194108.
[20] IDROBO J C,  S, NEMETH K, JELLINEK J, FERRANDO R. First-principles isomer-specific absorption spectra of Ag11 [J]. Physical Review B, 2007, 75(23): 233411.
S, NEMETH K, JELLINEK J, FERRANDO R. First-principles isomer-specific absorption spectra of Ag11 [J]. Physical Review B, 2007, 75(23): 233411.
[21] LI Wei-yin, CHEN Fu-yi. Effect of multiple exciton generation on ultraviolet–visible absorption of Ag–Cu clusters: Ab initio study [J]. Journal of Alloys and Compounds, 2014, 607: 238–244.
[22] MA Wen-qiang, CHEN Fu-yi. Optical and electronic properties of Cu doped Ag clusters [J]. Journal of Alloys and Compounds, 2012, 541: 79–83.
[23] TAFOUGHALT M A, SAMAH M. Density functional investigation of structural and electronic properties of small bimetallic silver–gold clusters [J]. Physica B: Condensed Matter, 2012, 407(12): 2014–2024.
[24] TIAN Dong-xu, ZHANG Hua-lei, ZHAO Ji-jun. Structure and structural evolution of (n=3–22) clusters using a genetic algorithm and density functional theory method [J]. Solid State Communications, 2007, 144(3): 174–179.
[25] PEREIRO M, BALDOMIR D. Structure of small silver clusters and static response to an external electric field [J]. Physical Review A, 2007, 75(3): 033202.
[26] LI Hai-sheng, JI Yong, WANG Fei, LI S F, SUN Q, JIA Yu. Ab initio study of larger Pbn clusters stabilized by Pb7 units possessing significant covalent bonding [J]. Physical Review B, 2011, 83(7): 075429.
[27] KONG Xiang-feng, YANG Bin, XIONG Heng, KONG Lin-xin, LIU Da-chun, XU Bao-qiang. Thermodynamics of removing impurities from crude lead by vacuum distillation refining [J]. Transactions of Nonferrous Metals Society of China, 2014, 24(6): 1946–1950.
[28] SONG Bing-yi, JIANG Wen-long, YANG Bin, XU Bao-qiang, YANG Qi-tong, XU Shuai, LIU Da-chun. Investigation on recycling of Ag from Pb-Cu-Ag Alloy by vacuum distillation [C]// Energy Technology 2015: Carbon Dioxide Management and Other Technologies: TMS 2015 144th Annual Meeting & Exhibition. Orlando: TMS, 2015: 181–191.
[29] WANG Bao-lin, ZHAO Ji-jun, CHEN Xiao-shuang, SHI Da-ning, WANG Guang-hou. Atomic structures and covalent-to-metallic transition of lead clusters Pbn (n=2–22) [J]. Physical Review A, 2005, 71(3): 033201.
[30] RAJESH C, MAJUMDER C. Atomic and electronic structures of neutral and charged Pbn clusters (n=2–15): Theoretical investigation based on density functional theory [J]. The Journal of Chemical Physics, 2007, 126(24): 244704.
[31] DUAN Shao-fei, CHEN Xiu-min, YANG Bin, YU Qing-chun, Xu Bao-qiang, LIU Da-chun. Calculation of interaction of AlCl, AlCl2 and AlCl3 on Al4C3 (001) Al4CO4 (001) and Al2CO (001) planes [J]. Journal of Central South University, 2015, 22(1): 43–58.
[32] LU Yong, ZHOU Yue-zhen, Chen Xiu-min, LI Zi-yong, YU Qing-chun, LIU Da-chun, YANG Bin, XU Bao-qiang. Thermodynamic analysis and dynamics simulation on reaction of Al2O and AlCl2 with carbon under vacuum [J]. Journal of Central South University, 2016, 23(2): 286–292.
[33] YUAN H Y, KUANG A L, TIAN C L, CHEN H. Structural and electronic properties of Aun-xPtx (n=2–14; x≤n) clusters: The density functional theory investigation [J]. AIP Advances, 2014, 4(3): 037107.
[34] JACHSCHATH C, RABIN I, SCHULZE W. Electron impact ionization of silver clusters Agn, n≤36 [J]. Zeitschrift für Physik D: Atoms, Molecules and Clusters, 1992, 22(2): 517–520.
[35] SAITO Y, YAMAUCHI K, MIHAMA K, NODA T. Formation and ionization potentials of lead clusters [J]. Japanese Journal of Applied Physics, 1982, 21(6A): L396.
[36] PARGA J R, VALENZUELA J L, MUZQUIZ G G, OJEBUOBOH F K. Recycling lead to recover refractory precious metals [J]. JOM, 2001, 53(12): 19–21.
[37] KUANG Xiang-jun, WANG Xin-qiang, LIU Gao-bin. A density functional study on the AunAg (n=1–12) alloy clusters [J]. Journal of Alloys and Compounds, 2013, 570: 46–56.
[38] DE BAS B S, FORD M J, CORTIE M B. Low energy structures of gold nanoclusters in the size range 3–38 atoms [J]. Journal of Molecular Structure: Theochem, 2004, 686(1): 193–205.
[39] SENZ V, FISCHER T, OEΙ NER P, TIGGESBUMKER J, STANZEΙ J, BOSTEDT C, THOMAS H, SCHFFLER M, FOUCAR L, MARTINS M, NEVILLE J, NEEB M, MΙΙER T, WURTH W, R
NER P, TIGGESBUMKER J, STANZEΙ J, BOSTEDT C, THOMAS H, SCHFFLER M, FOUCAR L, MARTINS M, NEVILLE J, NEEB M, MΙΙER T, WURTH W, R HΙ E, DRNER R, SCHMIDT-BCKING H, EBERHARDT W, GANTEFR G, TREUSCH R, RADCLIFFE P, MEIWES-BROER K H. Core-hole screening as a probe for a metal-to-nonmetal transition in lead clusters [J]. Physical Review Letters, 2009, 102(13): 138303.
HΙ E, DRNER R, SCHMIDT-BCKING H, EBERHARDT W, GANTEFR G, TREUSCH R, RADCLIFFE P, MEIWES-BROER K H. Core-hole screening as a probe for a metal-to-nonmetal transition in lead clusters [J]. Physical Review Letters, 2009, 102(13): 138303.
(Edited by HE Yun-bin)
中文导读
采用密度泛函理论对二元合金PbnAgn (n=2–12)团簇的结构和电子性质进行研究
摘要:采用密度泛函理论中的GGA-BLYP近似对PbnAgn (n=2–12)团簇的结构和电子性质进行研究,且所有的计算都是在DMol3模块中进行的。采用优化过的PbnAgn (n=2–12)团簇作为从头算分子动力学的初始结构并采用从头算分子动力学对其基态结构进行搜索,最终得到了PbnAgn (n=2–12)团簇的基态结构。基于PbnAgn (n=2–12)团簇的基态结构演变规律发现,Ag原子倾向于聚集在一起且占据在团簇的中心的位置而被周围的Pb原子包围,这也与铅富集贵金属的现象以及文献报道的实验结果一致。对PbnAgn (n=2–12)团簇的平均结合能、HOMO-LUMO能隙、垂直电子电离势、垂直电子亲和势、化学硬度η、HOMO轨道、LUMO轨道和态密度进行了计算。研究结果表明,当n≤5时,HOMO-LUMO能隙、垂直电子电离势、垂直电子亲和势和化学硬度η的值显示出了明显的奇–偶振荡现象;随着团簇尺寸的增大,PbnAgn (n=2–12)团簇的化学稳定性降低,且显示出由绝缘性向金属性的过渡;PbnAgn (n≥9)团簇是用于计算Pb–Ag合金性质的理想候选者。这些可很好地用铅原子和银原子在–0.5 Ha~0.25 Ha之间的态密度(DOS)来解释。
关键词:密度泛函理论;PbnAgn (n=2–12)团簇;从头算分子动力学;基态结构;合金
Foundation item: Project(51664032) supported by the Regional Foundation of the National Natural Science Foundation of China; Project(51474116) supported by the General Program of the National Natural Science Foundation of China; Project(U1502271) supported by the Joint Foundation of the NSFC-Yunnan Province, China; Project(2014HA003) supported by the Cultivating Plan Program for the Leader in Science and Technology of Yunnan Province, China; Project(2014RA4018) supported by the Program for Nonferrous Metals Vacuum Metallurgy Innovation Team of Ministry of Science and Technology, China; Project(2016YFC0400404) supported by the National Key Research and Development Program of China; Project(51504115) supported by the Youth Program of National Natural Science Foundation of China; Project(IRT_17R48) supported by the Program for Innovative Research Team in University of Ministry of Education of China
Received date: 2016-09-18; Accepted date: 2016-12-14
Corresponding author: CHEN Xiu-min, PhD, Professor; Tel: +86-871-5107208; E-mail: chenxiumin9@hotmail.com; ORCID: 0000- 0003-1660-6106

