

![]()

Trans. Nonferrous Met. Soc. China 22(2012) 1437-1444
First principles calculation of intermetallic compounds in FeTiCoNiVCrMnCuAl system high entropy alloy
NONG Zhi-sheng1,2, ZHU Jing-chuan1,2, YU Hai-ling1, LAI Zhong-hong1
1. School of Materials Science and Engineering, Harbin Institute of Technology, Harbin 150001, China;
2. National Key Laboratory for Precision Hot Processing of Metals,
Harbin Institute of Technology, Harbin 150001, China
Received 17 June 2011; accepted 21 October 2011
Abstract:
The structural, electronic and elastic properties of common intermetallic compounds in FeTiCoNiVCrMnCuAl system high entropy alloy were investigated by the first principles calculation. The calculation results of formation enthalpy and cohesive energy show that FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases may form in the formation process of the alloy. Further studies show that FeTi, Fe2Ti, AlCrFe2, Co2Ti and AlMn2V phases with higher shear modulus and elastic modulus would be excellent strengthening phases in high entropy alloy and would improve the hardness of the alloy. In addition, the partial density of states was investigated for revealing the bonding mode, and the analyses on the strength of p-d hybridization also reveal the underlying mechanism for the elastic properties of these compounds.
Key words:
FeTiCoNiVCrMnCuAl system; high entropy alloy; first principles calculation; phase stability;
1 Introduction
High entropy (HE) alloy is a new field of alloys. It contains at least five major elements, and the concentration for each element ranges between 5% and 35% [1,2]. Unlike traditional concept, HE alloy can form simple solid solutions, and it possesses many excellent properties, such as high hardness, high strength and good wear resistance [3-6]. However, the formation of intermetallic compounds may occur due to multiple principal elements in HE alloy. As the amount of alloying elements increases, the probability of intermetallic compounds appearance also increases. The basic mechanical properties of the alloy would be affected by intermetallic compounds formed in HE alloy, which depends on the types and characteristics of the compounds. WANG et al [7] investigated the crystal structure of CoCrCuFeNiTix (x=0, 0.5, 0.8 and 1.0) HE alloys using X-ray diffraction, and found that there was Fe2Ti phase existing when the content of Ti was 0.8 and 1.0, and the plasticity of these alloys was affected by the appearance of Fe2Ti phase. ZHANG et al [8] reported that FeTi phase existing in CoCrFeNiTiAl0.5 HE alloy played an important role in the strength of the alloy. Generally speaking, the mechanical properties of materials are affected by the structural, electronic and elastic properties of intermetallic compounds which appear in the matrix of materials to a certain extent. The difficulty degree of formation and the stability of crystal structure play important parts in the appearance of intermetallic compounds. Recently, theoretical calculations based on density functional theory (DFT) have successfully been used to evaluate the stability and predict the relevant properties of intermetallic compounds [9-11]. Moreover, the elastic constants can also provide information on the phase stability of materials and play an important role in determining the mechanical properties of materials [12,13].
In order to fully understand the influence of intermetallic compounds on HE alloy, it is necessary to investigate the structural, electronic and elastic properties of the intermetallic compounds systematically by theoretical methods. However, to the best of our knowledge, there is no theoretical study reported from the first principles calculation in this field, because the amount of intermetallic compounds with the possibility of formation is large and the theoretical conclusions require a great deal of further experimental research on these compounds as a proof.
In this study, a systematic investigation was carried out on the lattice constants, formation enthalpy, cohesive energy, density of states (DOS) and elastic constants of common intermetallic compounds in HE alloy by first principles calculations. FeTiCoNiVCrMnCuAl virtual system is chosen for research because these nine elements are the most common components of high entropy alloy [1, 4-6]. The present study would provide useful results regarding structural, electronic and elastic properties of intermetallic compounds in the FeTiCoNiVCrMnCuAl system HE alloy for the analysis of mechanical properties and for future characterization of relevant properties of high entropy alloy.
2 Calculation method
For theoretical analyses on the structural, electronic and elastic properties of intermetallic compounds in FeTiCoNiVCrMnCuAl system high entropy alloy, a Cambridge Serial Total Energy Package (CASTEP) was used for the first principles calculation. This software package employed the first principles plane wave pseudo-potential method based on the density functional theory (DFT) [14,15], in which the exchange and correlation terms were described with the generalized gradient approximation (GGA) of Perdew Burke Ernzerhof (PBE) parameterized by PERDEW [16,17]. Ultrasoft pseudo-potential was used to describe electron-ion interaction. The cutoff energies were all set at 350 eV for these calculations under a series of tests. Each calculation was considered converged when the maximum force on the atom was below 0.01 eV/?, the maximum displacement between cycles was below 5.0��10-4 ?, the maximum stress was below 0.02 GPa and the energy change was below 5.0��10-6 eV/atom.
As known, there is a large number of intermetallic compounds with different components and structures based on the FeTiCoNiVCrMnCuAl system. However, some of them with complex structure are unusual. Previous studies reported the information about the structure of most common binary and ternary intermetallic compounds based on FeTiCoNiV- CrMnCuAl system such as AlCo [18] , AlNi [19] , AlNi2Ti [20] and FeTi [21], and the space groups and corresponding lattice constants of these compounds are listed in Table 1 [22-43]. Therefore, these 38 common compounds were chosen for calculation in this work. In the calculation section, the formation enthalpy, cohesive energy, lattice constants, electronic and elastic properties of these 38 intermetallic compounds were investigated by the first principles calculation using the software of Materials Studio 4.0.
3 Results and discussion
3.1 Formation enthalpy and cohesive energy
From the view of thermodynamics, a lower formation enthalpy defined as the total energy difference between the compound and its constituents in proportion to the composition means better forming ability. The formation enthalpy of single metal atom Hf is calculated as [44,45]:
![]() (1)
(1)
where Etot is the total energy of compounds; n (or m) is the atom number of A (or B) element in a unit cell; and ![]() (or
(or ![]() ) is the energy per atom of A (or B) element in the solid state of the crystal structure. In this study, the formation enthalpy of single metal atom Hf is used for measuring the forming ability of intermetallic compounds.
) is the energy per atom of A (or B) element in the solid state of the crystal structure. In this study, the formation enthalpy of single metal atom Hf is used for measuring the forming ability of intermetallic compounds.
Similarly, a lower cohesive energy means better structural stability. Cohesive energy Ec is defined as the work to decompose the crystal into single free atoms and it is expressed as:
![]() (2)
(2)
where ![]() and
and ![]() are the energy per atom of A and B under isolated state, respectively. The cohesive energy of single metal atom Ec is used for measuring the stability of intermetallic compounds.
are the energy per atom of A and B under isolated state, respectively. The cohesive energy of single metal atom Ec is used for measuring the stability of intermetallic compounds.
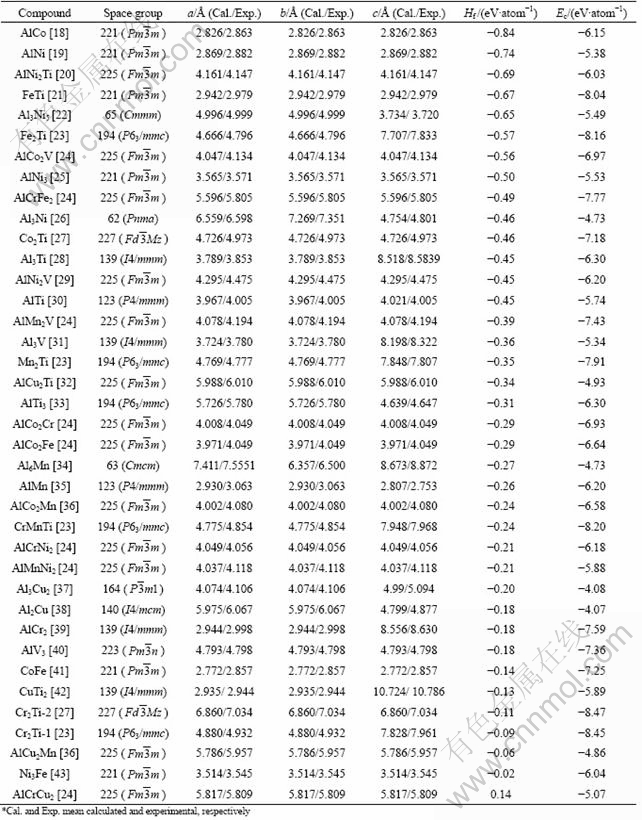
The calculation results of formation enthalpy Hf, cohesive energy Ec and lattice constants for these 38 intermetallic compounds are listed in Table 1. The lattice constants of compounds are calculated by geometrical optimization, and the formation enthalpy Hf is calculated by Eq. (1), cohesive energy Ec is calculated by Eq. (2). The results are all calculated at 0 K (including lattice constants, elastic constants and density of states). It can be seen that the calculated lattice constants match fairly closely the corresponding experimental ones with error of less than 3%, which indicates that these parameters are reasonable for this calculation. If thermodynamic effects are considered, the calculation results should be in good agreement with the experimental data. All the formation enthalpies of the compounds are negative except one of AlCrCu2 phase, which means these intermetallic compounds would form [46]. The formation enthalpy Hf of AlCo phase with the value of -0.84 eV/atom is lower than the others, which indicates that the AlCo phase would form more easily than the others at 0 K. There is a relative sequence of the forming ability from up to down, as listed in Table 1. Theoretically, AlCo, AlNi, AlNi2Ti, FeTi, Al3Ni5 and Fe2Ti phases with the value of formation enthalpy less than -0.35 eV/atom would form easily in the formation process of FeTiCoNiVCrMnCuAl system HE alloy. However, not all of these intermetallic compounds would stably exist in this HE alloy. FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases with the value of cohesive energy less than -7.0 eV/atom have a more stable crystal structure than other compounds. Although some compounds with lower formation enthalpy would form easily, the structures of these compounds are not stable enough. Therefore, FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases with low formation enthalpy and cohesive energy would form in the formation process of FeTiCoNiVCrMnCuAl system HE alloy in the view of thermodynamics, which agrees with the experimental results of previous studies. There was FeTi intermetallic compounds forming in CoCrFeNiTiAlx HE alloys when the content of Al was 0.5 [8]. For CoCrFeNiTiAl0.5 HE alloy, which belongs to CoCrFeNiTiAl system, FeTi, Fe2Ti, AlCrFe2 and Co2Ti phases may form in this alloy based on the results of calculation. Although there is lack of related experimental data about the appearance of AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases in HE alloy, the results still can be used as a guide for predicting the appearance of intermetallic compounds in HE alloy. In addition, the formation enthalpy and cohesive energy of copper compounds are not low enough and they would not form, which is due to the reason that copper element is almost rich in interdendritic region and is separated with other elements [4-6].
Table 1 Lattice constant, formation enthalpy and cohesive energy of intermetallic compounds
3.2 Elastic properties
For a stable crystal structure, its elastic constants Cij should satisfy the Born stability criteria [47]. The mechanical stability of the crystal implies that the strain energy must be positive. The stability criteria for cubic and hexagonal crystal are given as:
![]() ,
, ![]() ,
, ![]() ,
,![]() (3)
(3)
![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
,
![]() (4)
(4)
The theoretical elastic constants Cij for FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases are listed in Table 2. It can be seen that these calculated elastic constants Cij satisfy the Born stability criteria, suggesting that FeTi, AlCrFe2, Co2Ti and AlMn2V with cubic structure and Fe2Ti and Mn2Ti with hexagonal structure are mechanically stable.
The mechanical properties of intermetallic compounds play an important role on the mechanical properties of HE alloy. From the calculated elastic constants, other structural properties such as bulk modulus (B), shear modulus (G) and elastic modulus (E) can be derived using Voigt-Reuss-Hill (VRH) approximation. The final values can be taken as [48]:
B=(BV+BR)/2; G=(GV+GR)/2; E=9BG/(3B+G) (5)
where V and R refer to the models of Voigt and Reuss, respectively.
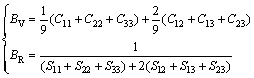 (6)
(6)
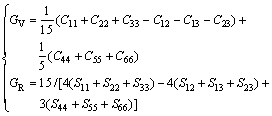 (7)
(7)
where Cij and Sij are elastic constants and elastic compliances, respectively; Sij is the inverse matrix of Cij. B, G and E are calculated by Eqs. (5)-(7), respectively.
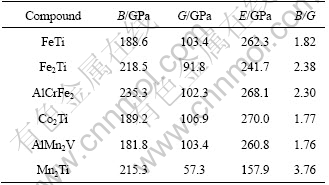
The theoretical elastic constants of FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases which would form in the formation process of FeTiCoNiVCrMnCuAl system HE alloy are listed in Table 3. It can be seen that FeTi, Fe2Ti, AlCrFe2, Co2Ti and AlMn2V phases with higher shear modulus G and elastic modulus E present better rigidity than Mn2Ti phase. Therefore, FeTi, Fe2Ti, AlCrFe2, Co2Ti and AlMn2V phases would be the strengthening phases in FeTiCoNiVCrMnCuAl system HE alloy, and the addition of Fe, Al and Ti elements could improve the hardness of HE alloy. The ratio of bulk to shear modulus B/G was proposed by PUGH [49] to predict the brittle or ductile behaviors of materials. If B/G>1.75, ductile behavior can be predicted for this material, otherwise the material shows a brittle behavior. As listed in Table 3, Mn2Ti, AlCrFe2 and Fe2Ti phases present good ductile manner, and FeTi, Co2Ti and AlMn2V phases have a little ductile manner. Mn2Ti and Fe2Ti show good ductile manner probably due to their particular C14 hexagonal structure.
Besides B/G, C11-C12 and elastic modulus E are another two important parameters for the mechanical properties of materials [50]. The smaller values of C11-C12 and elastic modulus E correspond to better plasticity of materials. The calculated results in Tables 2 and 3 show that Mn2Ti has lower values of C11-C12 and elastic modulus E, implying a better plasticity.
Table 2 Calculated elastic constants Cij of intermetallic compounds
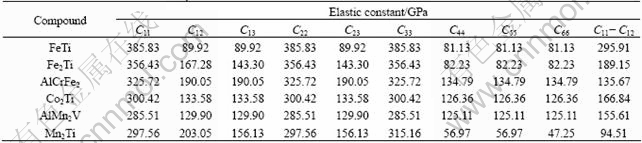
Table 3 Calculated bulk modulus B, shear modulus G and elastic modulus E of intermetallic compounds
3.3 Electronic structure
The density of states (DOS) is an important theoretical quantity to reveal the nature of chemical bonding interactions within compounds and understand their structural stability mechanism. The total and partial DOS at equilibrium lattice constants including FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases are calculated, as shown in Fig. 1. The two large occupied peaks are located at energy range between -4 eV and 4 eV, and the Fermi energy is about in the middle of them. The values of total DOS of these compounds at Fermi level N(Ef) are all higher than 0, which indicates that these compounds present obvious metallic behavior. Since the details of the partial density of states (PDOS) of binary compounds FeTi, Fe2Ti, Co2Ti and Mn2Ti are similar, FeTi phase is chosen as a prototype. Similarly, AlCrFe2 phase is chosen for the example of ternary compounds. From Fig. 1(a), it is found that the main bonding peaks between -10 and -5 eV mainly originate from the contribution of valence electron numbers of Fe(s) and Ti(s) orbits, the main bonding peaks between -5 and 5 eV are the result of the bonding Fe(d) and Ti(d) hybridized with little Fe(p) and Ti(p), implying that the covalent-like bonding exists in FeTi phase. The main bonding peaks between 5 and 20 eV are the contribution of Fe(s), Ti(s), Fe(p) and Ti(p) (It is absent in Fe2Ti, Co2Ti and Mn2Ti phases). From Fig. 1(c), the main bonding peaks of AlCrFe2 are located at three energy ranges, between -10 and -6 eV are dominated by the valence electron numbers of Al(s), Fe(s) and Fe(p) orbits; between -6 and 5 eV are the contribution of Cr(d) and Fe(d) hybridized with slight Al(p); between 5 and 15 eV are the contribution of Al(p), Al(s) and Fe(p). The appearance of hybridization between Cr(d), Fe(d) and Al(p) also means the covalent-like bonding behavior in AlCrFe2 phase.
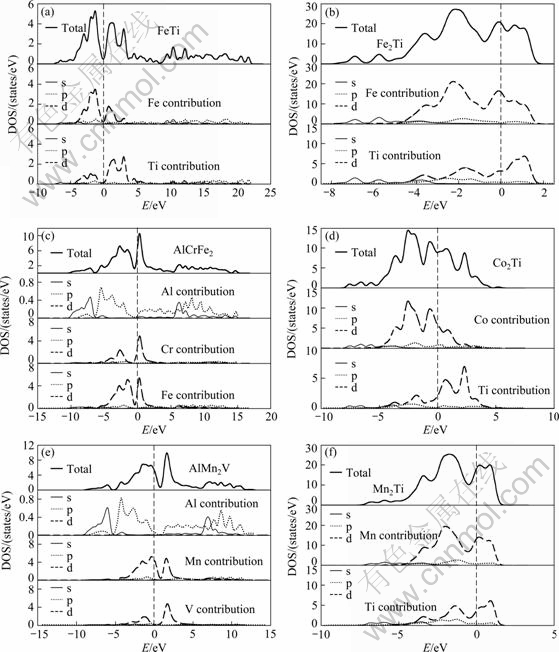
Fig. 1 Calculated total and partial densities of states of intermetallic compounds in FeTiCrCoNiVMnCuAl high entropy alloy (Fermi level is set to be 0 eV): (a) FeTi; (b) Fe2Ti; (c) AlCrFe2; (d) Co2Ti; (e) AlMn2V; (f) Mn2Ti
To further reveal the underlying mechanism for elastic properties, the electronic structure of these compounds is also discussed. The localization of bonding electrons which causes the hybridization is the dominant factor in governing the brittleness of the materials [51]. From Fig. 1, it can be seen that the strength of p-d hybridization resulting from Ti(p), Fe(p) and Ti(d) in FeTi phase (Co(p) and Ti(d) in Co2Ti phase; Al(p), Mn(d) and V(d) in AlMn2V phase) is higher than that in Fe2Ti, AlCrFe2 and Mn2Ti phases, suggesting that the brittle behavior is more evident in FeTi, Co2Ti and AlMn2V phases. These results of DOS are in agreement with the conclusions of elastic constants.
4 Conclusions
1) The formation enthalpy, cohesive energy, electronic structure and elastic constants of compounds in FeTiCoNiVCrMnCuAl system high entropy alloy are calculated and discussed by the first principles calculation based on density functional theory.
2) The results show that FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases with the value of formation enthalpy less than -0.35 eV/atom and the value of cohesive energy less than -7.0 eV/atom have a more stable crystal structure, thus would form in the formation process of FeTiCoNiVCrMnCuAl system alloy under the calculation results of formation enthalpy and cohesive energy.
3) The calculated theoretical elastic properties of FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases indicate that FeTi, Fe2Ti, AlCrFe2, Co2Ti and AlMn2V phases with higher shear modulus G and elastic modulus E would improve the hardness of high entropy alloy. In further studies, the calculation results of DOS for these 6 compounds reveal the bonding ability of compounds.
References
[1] YEH J W. Recent progress in high-entropy alloys [J]. Ann Chim Sci Mat, 2006, 31: 633-648.
[2] YEH J W, CHEN S K, LIN S J, GAN J Y, CHIN T S, SHUN T T, TSAU C H, CHANG S Y. Nanostructured high entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes [J]. Adv Eng Mater, 2004, 6: 299-303.
[3] HUANG P K, YEH J W, SHUN T T, CHEN S K. Multi principal element alloys with improved oxidation and wear resistance for thermal spray coating [J]. Adv Eng Mater, 2004, 6: 74-78.
[4] HSU C Y, YEH J W, CHEN S K, SHUN T T. Wear resistance and high temperature compression strength of FCC CuCoNiCrAl0.5Fe alloy with boron addition [J]. Metall Mater Trans A, 2004, 35: 1465-1469.
[5] TONG J, CHEN S K, YEH J W, SHUN T T, TSAU C H, LIN S J, CHANG S Y. Mechanical performance of the AlxCoCrCuFeNi high entropy alloy system with multiprincipal elements [J]. Metall Mater Trans A, 2005, 36: 1263-1271.
[6] YEH J W, CHEN S K, GAN J Y, LIN S J, CHIN T S, SHUN T T, TSAU C H, CHANG S Y. Formation of simple crystal structures in CuCoNiCrAlFeTiV alloys with multiprincipal metallic elements [J]. Metall Mater Trans A, 2004, 35: 2533-2536.
[7] WANG X F, ZHANG Y, QIAO Y, CHEN G L. Novel microstructure and properties of multicomponent CoCrCuFeNiTix alloys [J]. Intermetallics, 2007, 15: 357-362.
[8] ZHANG K B, FU Z Y, ZHANG J Y, WANG W M, WANG H, WANG Y C, ZHANG Q J, SHI J. Microstructure and mechanical properties of CoCrFeNiTiAlx high-entropy alloys [J]. Mat Sci Eng A, 2009, 508: 214-219.
[9] PENG J Z, WANG Y F, GRAY M F. First-principles study of structural stabilities and electronic properties of Mg-Nd intermetallic compounds [J]. Physica B, 2008, 403: 2344-2348.
[10] LIN L, LIANG P, YANG L, CHEN L J, LIU Z, WANG Y M. Phase stability comparison by first principle calculation and experimental observation of microstructure evolution in a Mg-6Gd-2Zn (wt%) alloy [J]. Mat Sci Eng A, 2010, 527: 2643-2648.
[11] YANG Y J, TAO X M, ZHU W J, LONG Z H, LIU H S, JIN Z P. First-principles calculation assisted thermodynamic modeling of Ti-Co-Cu ternary system [J]. J Mater Sci Technol, 2010, 26: 317-326.
[12] WU M M, TANG B Y, PENG L M, DING W J. Elastic and electronic properties of ScMn2 from ?rst-principles calculations [J]. Physica B, 2010, 405: 4812-4817.
[13] RAVINDRAN P, FAST L, KORZHAVYI P A, JOHANSSON B J. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2 [J]. J Appl Phys, 1998, 84: 4891-4905.
[14] HOHENBERG P, KOHN W. Inhomogeneous electron gas [J]. Phys Rev B, 1964, 136: 384-389.
[15] KOHN W, SHAM L J. Self-consitent equations including exchange and correlation effects [J]. Phys Rev A, 1965, 140: 1133-1139.
[16] PERDEW J P, LEVY M. Extrema of the density functional of the energy-excited-states from the ground-state theory [J]. Phys Rev B, 1985, 31: 6264-6272.
[17] PERDEW J P. Density-functional approximation for the correlation-energy of the inhomogeneous electron-gas [J]. Phys Rev B, 1986, 33: 8822-8824.
[18] DUTCHAK Y I, CHEKH V G. High-temperature X-ray diffraction study of the lattice dynamics of the compounds AlCo and AlNi [J]. Russ J Phys Chem, 1981, 55: 2342-2345.
[19] GAYDOSH D J, JECH R W, TIRAN R H. Ductility in rapidly solidified NiAl [J]. J Mater Sci Lett, 1985, 4: 138-140.
[20] SRIDHARAN S, NOWOTNY H, WAYNE S F. Investigations within the quarternary system titanium-nickel-aluminum-carbon [J]. Monatshefte fuer Chemie, 1983, 114: 127-135.
[21] MELNYK G, TREMEL W. Mononuclear coordination compounds based on a novel chelating triazole ligand [J]. J Alloy Compd, 2003, 349: 164-171.
[22] ENAMI K, NENNO S. New ordered phase in tempered 63.8Ni-1Co-Al martensite [J]. Transactions of the Japan Institute of Metals, 1978, 19: 571-580.
[23] OHBA T, KITANO Y, KOMURA Y. The charge-density study of the laves phases [J]. Acta Crystallogr C, 1984, 40: 1-5.
[24] BUSCHOW KHJ, VAN Engen P G. Alloys with composition ![]() (0��x��50) were investigated employing electron probe microanalysis (EPMA) and X-ray powder diffraction (XPD) [J]. J Magn Magn Mater, 1981, 25: 90-96.
(0��x��50) were investigated employing electron probe microanalysis (EPMA) and X-ray powder diffraction (XPD) [J]. J Magn Magn Mater, 1981, 25: 90-96.
[25] MOHAN RAO P V, SURYANARAYANA S V, SATYANARAYANA M K, NAGENDER NAIDY S V. The high-temperature thermal expansion of Ni3Al measured by X-ray diffraction and dilation methods [J]. J Phys-Condens Mat, 1989, 1: 5357-5361.
[26] BRADLEY A J, TAYLOR A. The crystal structures of Ni2A13 and NiA13 [J]. Philos Mag, 1937, 23: 1049-1067.
[27] OHBA T, KITANNO Y, KOMURA Y. The charge-density study of the laves phases, MgZn2 and MgCu2 [J]. Acta Crystallogr C, 1984, 40: 1-5.
[28] NORBY P, CHRISTENSEN A N. Sodium alanate nanoparticles for hydrogen storage [J]. Acta Chem Scand, 1986, 40: 157-159.
[29] MARAZZA R, FERRO R, RAMBALDI G. Some phases in ternary alloys of titanium, zirconium, and hafnium, with a MgAgAs or AlCu2Mn type structure [J]. Journal of the Less-Common Metals, 1975, 39: 341-345.
[30] DUWEZ P, TAYLOR J L. Crystal structure of TiAl [J]. Trans AIME, 1952, 194: 70-71.
[31] MAAS J, BASTIN G, Van LOO F, METSELAAR R. On the texture in diffusion-grown layers of silicides and germanides with the FeB structure, MeX (Me=Ti, Zr; X=Si, Ge) or the ZrSi2 structure [J]. Z Metallkd, 1983, 74: 294-299.
[32] RAMAN A, SCHUBERT K. Microstructure, deformation and fracture characteristics of an Al67Ni8Ti25 intermetallic alloy [J]. Z Metallkd, 1965, 56: 99-104.
[33] PENALOZA V A, HOUSKA C R. Refinements on the X-ray intensities from Ti3-2Al [J]. An Congr Nac Metal, 1983, 3: 55-59.
[34] KONTIO A, COPPENS P. New study of the structure of MnAl6 [J]. Acta Crystallogr B, 1981, 37: 433-435.
[35] KOESTER W, WACHTEL E. Magnetic investigation of Al-Mn alloys containing more than 25% Mn [J]. Z Metallkd, 1960, 51: 271-280.
[36] SOLTYS J. X-ray diffraction research of the order-disorder transitions in the ternary heusler alloys B2MnAl (B=Cu, Ni, Co, Pd, Pt) [J]. Phys Status Solidi A, 1981, 66: 485-491.
[37] RAMACHANDRARAO P, LARIDJANI M. A metastable phase Al3Cu2 [J]. J Mater Sci, 1974, 9: 434-437.
[38] MEETSMA A, de Boer J L, VAN SMAALEN S. Refinement of the crystal structure of tetragonal Al2Cu [J]. J Solid State Chem, 1989, 83: 370-372.
[39] BRADLEY A J, LU S S. The crystal structures of Cr2Al and Cr5Al8 [J]. Zeitschrift fuer Kristallographie, 1937, 96: 20-37.
[40] KODESS B N. Some phases in ternary alloys of titanium, zirconium, and hafnium, with MgAgAs or AlCu2Mn type structure [J]. Phys Status Solidi A, 1971, 25: 1-15.
[41] BAYLISS P. Revised unit-cell dimensions, space group, and chemical formula of some metallic minerals [J]. Can mineral, 1990, 28: 751-755.
[42] MUELLER M H, KNOTT H W. Powder metallurgy and metal ceramics [J]. Transactions of the Metallurgical Society of Aime, 1963, 227: 674-678.
[43] AHMED Z, BEVAN J C. Awaruite, iridian awaruite, and a new Ru-Os-Ir-Ni-Fe alloy from the sakhakot-quila complex, malakand agency [J]. Mineral Mag, 1981, 44: 225-230.
[44] MEDVEDEVA M I, GORNOSTYREV Y N, NOVIKOV D L, MRYASOV O N, FREEMAN A J. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl [J]. Acta Mater, 1998, 46: 3433-3442.
[45] SAHU B R. Electronic structure and bonding of ultra-light LiMg [J]. Mater Sci Eng B, 1997, 49: 74-78.
[46] SONG Y, GUO Z X, YANG R, LI D. First principles study of site substitution of ternary elements in NiAl [J]. Acta Mater, 2001, 49: 1647-1654.
[47] BORN M, HUANG K. Dynamical theory of crystal lattices [M]. Oxford: Oxford University Press, 1954: 10-12.
[48] YAO H Z, OUYANG L Z, CHING W Y. Ab initio calculation of the elastic constants of ceramic crystals [J]. J Am Ceram Soc, 2007, 90: 3194-3204.
[49] PUGH S F. Relations between elastic moduli and plastic properties of polycrystalline pure metals [J]. Philos Mag, 1954, 45: 823-843.
[50] SUMER A, SMITH J F. The surface acoustic wave properties of lithium gallium oxide [J]. J Appl Phys, 1962, 33: 858-860.
[51] ZHANG H, SHANG S L, WANG Y, SAENGDEEJING A, CHEN L Q, LIU Z K. First-principles calculations of the elastic, phonon and thermodynamic properties of Al12Mg17 [J]. Acta Mater, 2010, 58: 4012-4018.
FeTiCoNiVCrMnCuAlϵ���غϽ���
�����仯����ĵ�һ��ԭ������
ũ����1,2���쾰��1,2���ں���1�����Һ�1
1. ��������ҵ��ѧ ���Ͽ�ѧϵ�������� 150001��
2. ��������ҵ��ѧ ���������ȼӹ����Ҽ��ص�ʵ���ң������� 150001
ժ Ҫ�����õ�һ��ԭ���о�FeTiCoNiVCrMnCuAlϵ���غϽ��г����Ľ����仯����Ľṹ�����Ӻ͵������ʡ��γ��ʺͽ���ܼ�����������FeTi��Fe2Ti��AlCrFe2��Co2Ti��AlMn2V �� Mn2Ti���п����ڸ��غϽ��γɵĹ����г��֡���һ���о����֣�FeTi��Fe2Ti��AlCrFe2��Co2Ti �� AlMn2V����нϸߵļ���ģ���͵���ģ�����ڸ��غϽ��п�����Ϊ��ǿ�����ӺϽ��ǿ�ȡ�ͨ���о��ֲ���̬�ܶ�����ʾ�м���ļ������ã����о�p-d�ӻ���ǿ�ȣ���ʾ�м���ĵ���������DZ�ڻ�����
�ؼ��ʣ�FeTiCoNiVCrMnCuAlϵ�����غϽ𣻵�һ��ԭ�����㣻���ȶ���
(Edited by FANG Jing-hua)
Foundation item: Project supported by the National Key Laboratory Opening Funding of Advanced Composites in Special Environments in Harbin Institute of Technology, China
Corresponding author: ZHU Jing-chuan; Tel: +86-451-86413792; Fax: +86-451-86413922; E-mail: zhujc1@gmail.com
DOI: 10.1016/S1003-6326(11)61338-1
Abstract: The structural, electronic and elastic properties of common intermetallic compounds in FeTiCoNiVCrMnCuAl system high entropy alloy were investigated by the first principles calculation. The calculation results of formation enthalpy and cohesive energy show that FeTi, Fe2Ti, AlCrFe2, Co2Ti, AlMn2V and Mn2Ti phases may form in the formation process of the alloy. Further studies show that FeTi, Fe2Ti, AlCrFe2, Co2Ti and AlMn2V phases with higher shear modulus and elastic modulus would be excellent strengthening phases in high entropy alloy and would improve the hardness of the alloy. In addition, the partial density of states was investigated for revealing the bonding mode, and the analyses on the strength of p-d hybridization also reveal the underlying mechanism for the elastic properties of these compounds.
[1] YEH J W. Recent progress in high-entropy alloys [J]. Ann Chim Sci Mat, 2006, 31: 633-648.
[14] HOHENBERG P, KOHN W. Inhomogeneous electron gas [J]. Phys Rev B, 1964, 136: 384-389.
[30] DUWEZ P, TAYLOR J L. Crystal structure of TiAl [J]. Trans AIME, 1952, 194: 70-71.
[37] RAMACHANDRARAO P, LARIDJANI M. A metastable phase Al3Cu2 [J]. J Mater Sci, 1974, 9: 434-437.
