
J. Cent. South Univ. (2016) 23: 2818-2826
DOI: 10.1007/s11771-016-3345-0
Analysis on differentially expressed genes in watermelon rind color based on RNA–Seq
YANG Kan-kan(杨侃侃)1, 2, LIANG Zhi-huai(梁志怀)2, WU Cai-jun(吴才君)1
1. College of Agronomy, Jiangxi Agricultural University, Nanchang 330045, China;
2. Hunan Watermelon and Muskmelon Institute, Hunan Academy of Agricultural Sciences, Changsha 410125, China
 Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Abstract:
In order to screen the genes controlling watermelon rind color and luster, the experiment was carried out with yellow watermelon skin mutants as tester and green wild type watermelon as control, and transcriptome sequencing and bioinformatics analysis were done. The results show that 34.27Gb clean data were got by transcriptome sequencing. There are 261 differentially expressed genes among Y1_vs_G1, Y2_vs_G2 and Y3_vs_G3. The pathways contenting most differentially expressed genes are plant hormone signal transduction pathway, phenylpropanoid biosynthesis pathway, photosynthesis pathway, starch and sucrose metabolism pathway. 9-cis-epoxycarotenoid dioxygenase (Cla002942), alcohol dehydrogenase (Cla004992), photosystem I reaction center subunit III, chloroplastic (precursor) (Cla009181), long-chain acyl coenzyme A synthetase (Cla017341), threonine dehydratase biosynthetic (Cla018352) candidates genes were screened out.
Key words:
watermelon;rind color;transcriptome sequencing;differentially expressed genes;
1 Introduction
Watermelon rind color is an important commodity in watermelon consumption characteristics in modern society; therefore, high quality, high yield and appearance for gorgeous watermelon varieties have a great significance to enrich watermelon varieties structure and improve the quality. The genetic of watermelon rind color and luster was analyzed by HUANG [1]. His results show that the rind appearing yellow is controlled by a dominant gene; yellow relative to green is as the dominant gene; watermelon rind color is genetic quality traits; watermelon rind yellow is controlled by a single dominant gene. The yellow rind, yellow petioles and yellow veins are controlled by the same gene, and they show the performance of different organs controlled by the same kind of anthocyanin. SZAMOSI et al [2] researched watermelon germplasm resources which come from Hungary and Turkey, the results found that watermelon rind yellow belongs to quality traits. COCHRAN [3] reported that cucumber rind white is recessive to the green. LAWRENCE and TODD [4] reported that yellow-green (yg) is recessive to the dark green cucumber peel, and is dominant to light green. STRONG [5] and TKACHENKO [6] reported respectively that cucumber peel dark green or luster is controlled by the D gene, and dark green as the dominant and the skin color consistency or motley is controlled by u gene, motley as dominant. LIN et al [7] studied the color and luster of genetics with dark green and red linen color in angular sponge gourd and the results show that the genetic of red linen color in angular sponge gourd is controlled by a dominant nuclear gene. YANG et al [8] orientated the muskmelon peel color and found that the peel color is controlled by a single gene and yellow-green peel to white peel is dominant character. Beijing Academy of Agriculture and Forestry Sciences, Shenzhen BGI and Cornell University, and other units published that watermelon genome was parsed successfully in nature genetics. The watermelon genome provided a number of resources for watermelon in basic biological research, germplasm resources to foster, disease resistance and genetic improvement. Watermelon fruit mature process includes many varieties such as the size, color, sugar, nutritional ingredient. Researchers, combining watermelon’s pulp and peel and the transcriptome sequencing data, found that the most varieties are cell wall synthesis and flavonoid metabolism in the mature progress of pulp and peel [9-10]. In the present work, the experiment is carried out on the differentially expressed genes with yellow watermelon rind mutants as tester and green wild type watermelon as control through the transcriptome sequencing technologies and sequence analysis.
2 Experimental
2.1 Materials
The experimental materials were the yellow homozygous mutant after six generations of selfing and selecting in 2002 for the breeding of new varieties of watermelon found in a parent materials when mature fruit skin showed yellow (the original was green). The materials were planted in horticulture planting base of Jiangxi Agricultural University. The rind and pulp of the fruit were separated and immediately frozen in liquid nitrogen wrapped in foil and hermetic bag and then stored at -80 °C for further research. The materials were got from the fruit in different periods and different colors, respectively. The yellow rind and green rind watermelon’s fruit-set period, enlargement period and maturity period, were named Y1, Y2, Y3, G1, G2 and G3.
2.2 Methods
PlantRNAzol kit was used to extract the RNA of Y1, Y2, Y3, G1, G2 and G3 in the rind, and detect the concentration of the RNA by ultraviolet spectrophoto- meter and agarose gel electrophoresis.
The eukaryotic mRNA was enriched by magnetic beads with Oligo (dT), adding the fragmentation buffer to interrupt mRNA in random. Based on mRNA templates, the first cDNA chain was synthesized by random hexamers. Then adding the buffer solution, dNTPs, RNase H and DNA polymerase I, the second cDNA chain was synthesized. cDNA was purified by AMPure XP beads. Repairing the end, adding A tail and connecting sequencing joint of the double-stranded cDNA which is purified, and fragment size was chosen by AMPure XP; finally, a cDNA library was got by PCR.
The quality of the library was inspected, and then HiSeq2500 was used for high-throughput sequencing the PCR products. Clean data were obtained by controlling raw data quality, and then compared with the reference genome. Based on comparison results, the transcriptome library quality evaluation, the SNP analysis and gene expression analysis were obtained.
On EBSeq, differentially expressed genes were obtained between the sample sets. FC (fold change)≥2 and FDR (false discovery rate) < 0.01 were the selection criteria. Fold change is the rate that is the amount of gene expression between the two samples (group). False discovery rate is a correction that uses the method of Benjamini-Hochberg to check the p-value which is got by the original hypothesis test. And the differentially expressed genes which are selected from the set were put into the database of Nr [11], GO [12] (gene ontology), COG [13] (cluster of orthologous groups), KEGG [14] (Kyoto Encyclopedia of genes and genomes) for the annotation information.
3 Results and analysis
3.1 Gene expression analysis
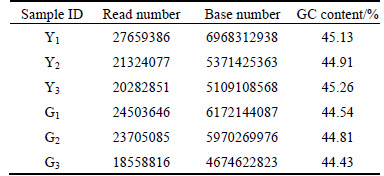
After the transcriptome sequencing of 6 groups of samples, there are 34.27 Gb clean data, and the average of clean data in each sample is 4.67 Gb. The Q30 values of samples Y1, Y2, Y3, G1, G2 and G3 are 87.51%, 87.65%, 87.16%, 88.21%, 88.85% and 86.85%, respectively. Q30 base is more 86.85% and the quality of transcriptome sequencing data is reliable (see Table 1).
Table 1 Sample sequencing data evaluation table
FPKM was used as an indicator to measure the level of gene expression, the length of the transcript and sequencing data normalization, and to eliminate the influence of gene length and sequencing depth on the data [15]. FPKM density contrast diagrams of sample are shown in Fig. 1.
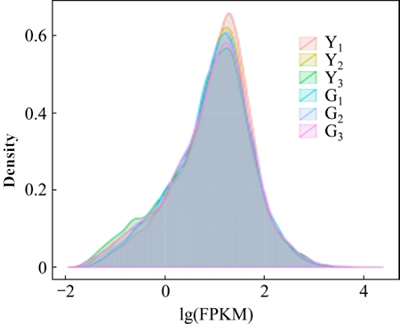
Fig. 1 FPKM density distribution of test materials (different colors represent different samples)
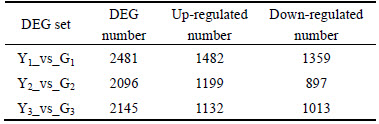
The tester and driver in fruit-set period were compared in enlargement period and maturity period. The results show that Y1_vs_G1 has 2481 differentially expressed genes; Y2_vs_G2 has 2096 differentially expressed genes; Y3_vs_G3 has 2145 differentially expressed genes. The numbers of differentially expressed genes are listed in Table 2.
Table 2 Statistics of number of differentially expressed genes
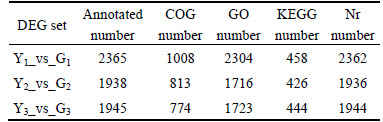
Each set of differentially expressed genes was annotated in COG, GO, KEGG and Nr. Each database annotation number of genes and the total numbers of comments are listed in Table 3.
Table 3 Numbers of annotated differentially expressed genes
Y1_vs_G1, Y2_vs_G2 and Y3_vs_G3 jointly own 261 differentially expressed genes. These genes have 164 up-regulate genes and 97 down-regulated genes. VEEN diagram of Y1_vs_G1, Y2_vs_G2 and Y3_vs_G3 jointly owning differentially expressed genes is shown in Fig. 2. Only 228 genes in these differentially expressed genes obtain annotations, and there are 140 up-regulated genes and 88 down-regulated genes.

Fig. 2 VEEN of differentially expressed genes
The common 228 differentially expressed genes of Y1_vs_G1, Y2_vs_G2 and Y3_vs_G3 were selected according to the condition of FDR < 0.001, log2FC≥4 or log2FC≤-4. The results are listed in Table 4.
3.2 COG annotation analysis on differentially expressed genes
COG functional annotation can make gene products classify the orthologous. In COG functional annotation, Y1_vs_G1 has 1008 differentially expressed genes, which are classified into 25 functions (see Fig. 3). The general function prediction only is the largest percentage, accounting for 29.66%. The second is transcription, accounting for 15.28%. Replication, recombination and repair account for 15.08%. Y2_vs_G2 has 813 differentially expressed genes which are classified into 25 functions, including general function prediction only for 19.05%, the transcription for 14.76%, amino acid transport and metabolism for 15.08%. There are 774 differentially expressed genes in Y3_vs_G3, including general function prediction only for 29.66%, the carbohydrate transport and metabolism for 12.66% and transcription for 15.28%.
3.3 GO annotation analysis on differentially expressed genes
GO analysis includes three parts: cellular component, molecular function and biological process. Y1_vs_G1 has a total of 2304 differentially expressed genes (see Fig. 4). In cellular component part, extracellular matrix part is the highest, accounting for about 45.45% and the second is extracellular region part, accounting for 27.69%. In molecular function part, the highest is structural molecule activity, accounting for 33.33%; receptor activity accounts for 19.33%. In biological processes part, the biggest is cell killing, accounting for 36.36%,; the next is growth, accounting for 18.82%. Y2_vs_G2 has a total of 1716 differentially expressed genes. One of the biggest differentially expressed genes in cellular component part is extracellular region part of 18.46%; the second is the extracellular matrix of 18.18%. In molecular function section, the largest percentage of differentially expressed is structural molecule activity of 33.33%; the next is antioxidant activity of about 20.25%. In biological process part, the largest is growth of 13.75%, followed by signaling of 12.98%. Y3_vs_G3 has a total of 1723 differentially expressed genes. In cellular component, one of the biggest differentially expressed genes is extracellular matrix part of 36.36%; the second is extracellular region part of about 23.08%. In molecular function section, the biggest difference is electron carrier activity of 19.30%, followed by the protein binding transcription factor activity of 16.67%. In biological process section, the biggest difference is locomotion, accounting for 22.78%.
3.4 KEGG annotation analysis on differentially expressed genes
KEGG functional annotation can analyze gene products metabolic pathways and function in the cell. In KEGG functional analysis of differentially expressed genes (see Fig. 5), Y1_vs_G1 has 458 differentially expressed genes annotated to 50 KEGG pathways, including 14 genes in organismal systems; genetic information processing has 40 genes; metabolism has 357 genes; cellular processes has 12 genes, environmental information processing has 35 genes. The pathway which has the most genes is plant hormone signal transduction, then photosynthesis and the last is starch and sucrose metabolism. Y2_vs_G2 has 426 differentially expressed genes in the annotation to the 50 KEGG pathways, including organism system with 10 genes. Genetic information processing has 25 genes; metabolism has 337 genes; cellular processes has 19 genes; environmental information processing has 35 genes; and the most pathway is plant hormone signal transduction, then phenylpropanoid biosynthesis, the last is starch and sucrose metabolism. Y3_vs_G3 has 444 differentially expressed genes in the annotation to the 50 KEGG pathways, including 16 organismal system genes. Genetic information processing has 50 genes; metabolism has 325 genes; cellular process has 14 genes; environmental information processing has 39 genes; and the most pathway is plant hormone signal transduction, followed by phenylpropanoid biosynthesis, and the last is photosynthesis.
Table 4 Main differentially expressed genes

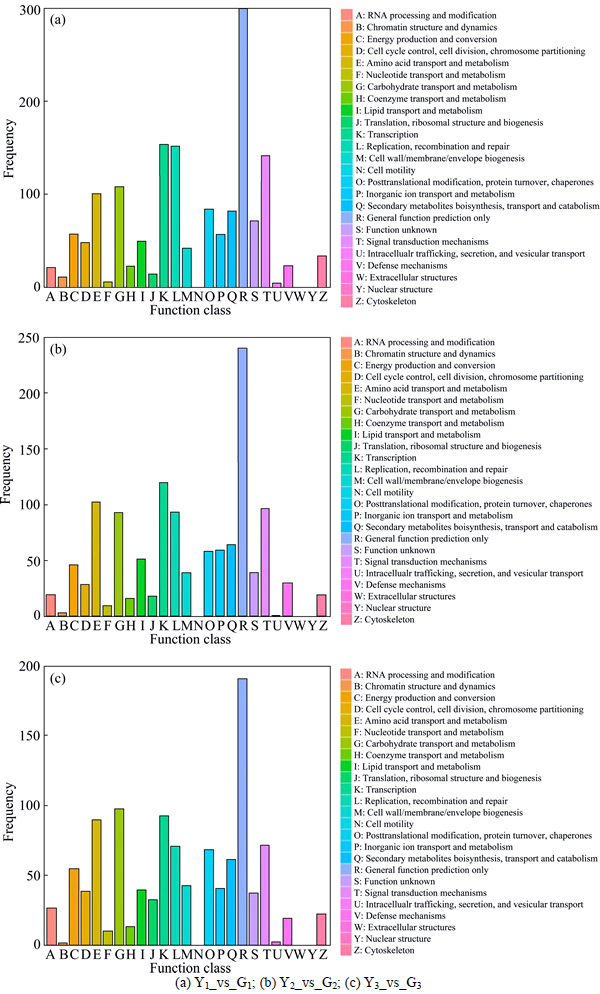
Fig. 3 COG annotation of differentially expressed genes:
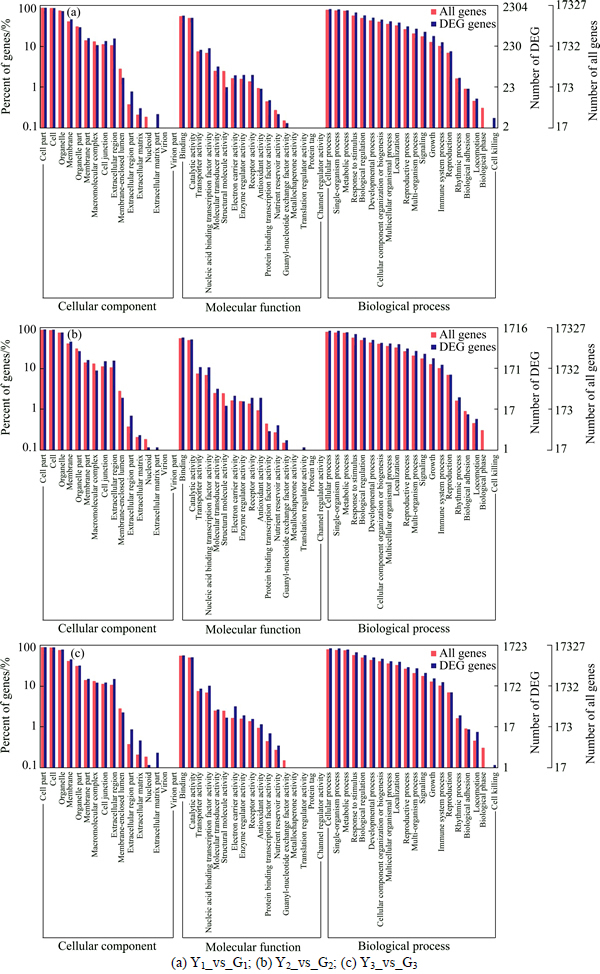
Fig. 4 GO annotation of differentially expressed genes:
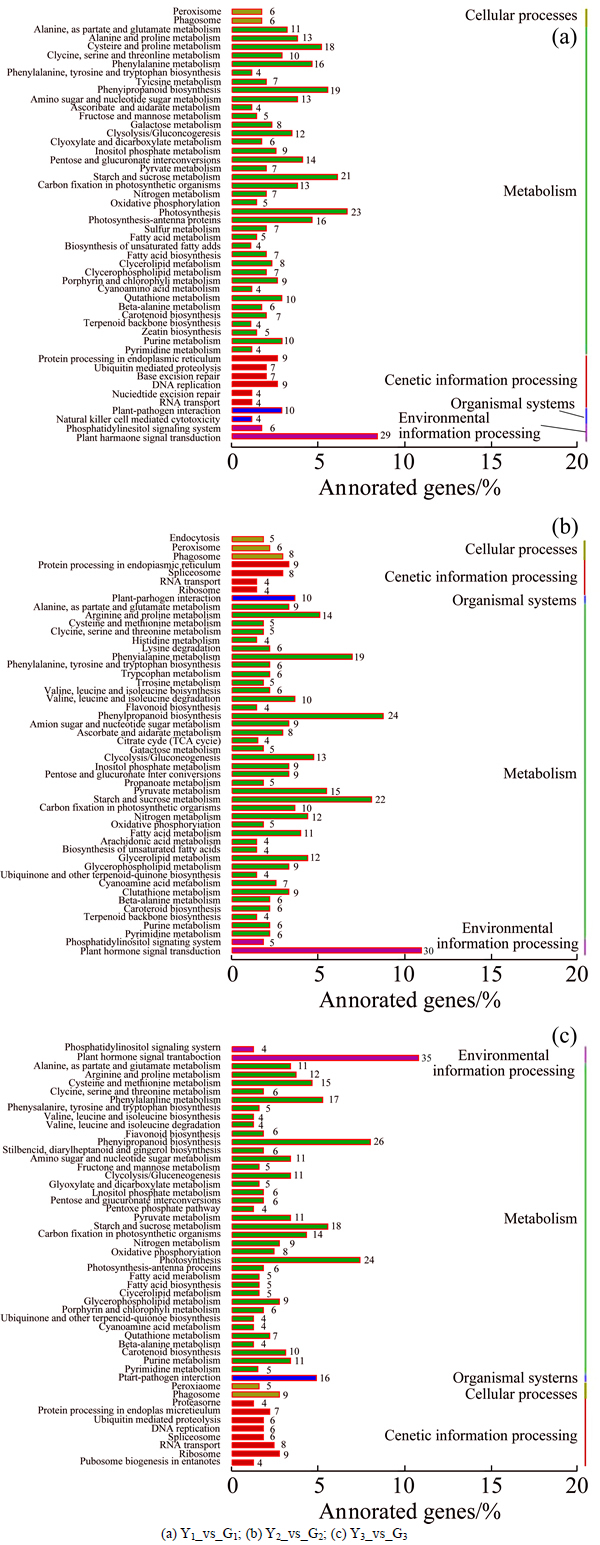
Fig. 5 KEGG annotation of differentially expressed genes:
4 Discussions
With watermelon mutant yellow rind color as tester and green wild watermelon rind as control, the test was sequencing based on RNA-Seq and bioinformatics analysis. The test selected out Y1_vs_G1, Y2_vs_G2, Y3_vs_G3 jointly owning a total of 261 differentially expressed genes, and COG, GO and KEGG functional annotation was simultaneously analyzed. According to the COG annotations, it was found that, in 25 categories, the number of gene general functions prediction only corresponds to the most commonly. According to GO functional annotation in three parts of 53 functions found in differentially expressed genes related to rind color, differentially expressed genes in cellular component are mainly classified into the extracellular matrix part; in molecular function, it is mainly classified into the structural molecule activity and enzyme regulator activity; in biological process, it is mainly classified into the growth and multi-organism process. According to KEGG function annotation of 50 pathways, it mainly annotates in plant hormone signal transduction of environmental information processing, phenylpropanoid biosynthesis, photosynthesis, starch and sucrose metabolism of metabolism.
The main pathways affecting watermelon rind color and luster are carotenoid synthesis, photosynthesis, metabolic pathway, biosynthesis of secondary metabolites, antibiotic biosynthesis, metabolism of drugs-cytochrome P450, chlorine substitutes and chloroform roalkene degradation and so on. Carotenoids synthetic mainly exists in the 9-cis-epoxycarotenoid dioxygenase (Cla002942), and it was reported in Arabidopsis thaliana, tobacco and rice involved in the carotenoids synthetic [16]. Abscisic acid biosynthesis is cracked into xanthoxin in plants by 9-cis lutein which is catalyzed by 9-cis-epoxycarotenoid dioxygenase, also the apoenzyme carotenoid composition which is oxidative chemical degradation by 9-cis-epoxycarotenoid dioxygenase, and it exists in many species, such as tomato recognition, bean, avocado, cowpea and Arabidopsis thaliana [17-18]. The gene which contents drug metabolism and cytochrome P450 and chlorinated and chloroform solvent roalkene degradation is alcohol dehydrogenase gene (Cla004992). The gene relating to photosynthesis is photosystem I reaction center subunit III, chloroplastic (Precursor) (Cla009181). The gene, which is located in the side of the inner cavity of thylakoid membrane, can be decomposed into complementary chlorophyll-protein complexes and contains chlorophyll, the combination of 47 kDa peptide CP2B [19-20]. Metabolic pathways have 9-cis- epoxycarotenoid dioxygenase (Cla002942), long-chain acyl coenzyme A synthetase (Cla017341), threonine dehydratase biosynthetic (Cla018352). Recently, the studies have shown that long-chain acyl coenzyme A fatty acid synthase (ACS) may participate in the compound of lipid synthesis and storage or tends towards acid oxidation [21]. In cell differentiation, a variety of hormones, nuclear transcription factor and the peroxidase can independently control sterol regulatory element binding protein-α (PPARα) [22-23]. Enzyme activity of threonine dehydrase is an important factor of the magnetic flux control in isoleucine biosynthesis, and the nitrogen remobilization of threonine dehydratase plays an important role in the aging of leaf [24-25].
5 Conclusions
This experiment bases on the transcriptome sequencing with the materials of yellow rind and green rind in three periods, and gets total numbers of 34.27 Gb clean data. Y1_vs_G1, Y2_vs_G2 and Y3_vs_G3 totally have 261 differentially expressed genes, in which up-regulate gene has 158, down-regulate gene has 58. Combining with COG annotation, GO annotation and KEGG annotation, 9-cis-epoxycarotenoid dioxygenase (Cla002942), alcohol dehydrogenase (Cla004992), photosystem I reaction center subunit III, chloroplastic (Precursor)(Cla009181), long-chain acyl coenzyme A synthetase (Cla017341), and threonine dehydratase biosynthetic (Cla018352) gene were selected out as candidates. The data of this experiment in the part of transcriptome sequencing on watermelon rind color and luster may provide data for watermelon genetic control and molecular breeding research.
References
[1] HUANG Shi-jie. The genetics of the watermelon rind yellow [J]. China Watermelon and Muskmelon, 1995(3): 15-16. (in Chinese)
[2] SZAMOSI C, SOLMAZ I, SARI N, BARSONY C. Morphological characterization of Hungarian and Turkish watermelon (Citrullus lanatus (Thunb.) Matsum. et Nakai) genetic resources [J]. Genetic Resources Crop Evolution, 2009, 56(8): 1091-1105.
[3] COCHRAN F D. Breeding cucumber for resistance to downy mildew [J]. Proceedings American Society for Horticultural Science, 1938, 35: 541-543.
[4] LAWRENCE K P, TODD C W. Review of genes and linkage groups in cucumber [J]. Hortscience, 1990, 25(6): 605-615.
[5] STRONG W J. Breeding experiments with the cucumber [J]. Scientia Agricola, 1931, 11: 333-346.
[6] TKACHENKO N N. Preliminary results of a genetic investigation of the cucumber, cucumissativusL [J]. Bul Applied Plant Breed, 1935, 2(9): 311-356.
[7] LIN Bao-ming, LIN Shi-sen. Prelimary study on the heredity of fruit length of angular sponge gourd [J]. Journal of South China Agricultural University, 2000, 21(2): 8-9. (in Chinese)
[8] YANG Guang-hua, FAN Rong, YANG Xiao-feng, HOU Jun-liang, YUAN Shi-chen, CAO Ming, WANG Xue-lin, LI Jing-song. Construction of a highly dense genetic map using SNP and mapping of three qualitative traints in Cucumis meb [J]. Acta Horticultural Sinica, 2014, 41(5): 898-906. (in Chinese)
[9] HOEN P A, ARIYUREK Y, THYGESEN H H, VREUGDENHI E, VOSSEN R H, MENEZES R X, BOER J M, OMMEN G J, DUNNEN J T. Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms [J]. Nucleic Acids Research, 2008, 36(21): e141.
[10] MURAT F, XU J H, TANNIER E, ABROUK M, GUILHOT N, PONT C, MESSING J, SALSE J. Ancestral grass karyotype reconstruction unravels new mechanisms of genome shuffling as a source of plant evolution [J]. Genome Research, 2010, 20: 1545-1557.
[11] DENG Yang-yang, LI Jian-qi, WU Song-feng, ZHU Yun-ping, CHEN Yao-wen, HE Fu-chu. Integrated NR database in protein annotation system and its localization [J]. Computer Engineering, 2006, 32(5): 71-74.
[12] ASHBURNER M, BALL C A, BLAKE J A, BOTSTEIN D, BUTLER H, CHERRY J M, DAVIS A P, DOLINSKI K, DWIGHT S S, EPPIG J T, HARRIS M A, HILL D P, ISSEL T L, KASARSKIS A, LEWIS S, MATESE J C, RICHARDSON J E, RINGWALD M, RUBIN G M, SHERLOCK G. Gene ontology: tool for the unification of biology [J]. Nature Genetics, 2000, 25(1): 25-29.
[13] TATUSOV R L, GALPERIN M Y, NATALE D A. The COG database: A tool for genome scale analysis of protein functions and evolution [J]. Nucleic Acids Research, 2000, 28(1): 33-36.
[14] KANEHISA M, GOTO S, KAWASHIMA S, OKUNO Y, HATTORI A M. The KEGG resource for deciphering the genome [J]. Nucleic Acids Research, 2004, 32: D277-D280.
[15] TRAPNELL C, WILLIAMS B A, PERTEA G, MORTAZAVI A, KWAN G, van BAREN M J, SALZBERG S L, WOLD B J, PACHTER L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation [J]. Nature Biotechnology, 2010, 28(5): 511-515.
[16] SATOSHI I, MASATOMO K, TERUAKI T, MASAAKI N, MOTOAKI S, TOMOHIKO K, SATOSHI T, YOSHITAKA K, KAZUKO Y, KAZUO S. Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis inarabidopsis[J]. The Plant Journal, 2001, 27(4): 325-333.
[17] QIN Xiao-qiang, JAN A D. Overexpression of a 9-cis- epoxycarotenoid dioxygenase gene in nicotiana plumbaginifolia increases abscisic acid and phaseic acid levels and enhances drought tolerance [J]. Plant Physiology February, 2002, 128(2): 544-551.
[18] XIA Hui, WU Shan, MA Feng-wang. Cloning and expression of two 9-cis-epoxycarotenoid dioxygenase genes during fruit development and under stress conditions from Malus [J]. Molecular Biology Reports, 2014, 41(10): 6795-6802.
[19] LUCA D, ROBERTO B, ALEXANDER R. Photoprotective mechanisms: Carotenoids [J]. Advances in Plant Biology, 2014, 5: 393-435.
[20] AKIHIKO Y, SAKAE K. A photoactive photosystem-II reaction-center complex lacking a chlorophyll-binding 40 kilodalton subunit from the thermophilic cyanobacterium Synechococcus sp [J]. Biochimica et Biophysica Acta, 1984, 765(2): 118-124.
[21] ROSALIND A C, TAL M L, CYNTHIA G. Do long-chain acyl-coa synthetasesregulate fatty acid entry into synthetic versus degradative pathways [J]. Nutrition, 2002, 132(8): 2123-2126.
[22] SOUPENE E, KUYPERS F A. Mammalian long-chain acyl-coa synthetases [J]. Experimental Biology Medicine, 2008, 233(5): 507-521.
[23] MOCKEL B, EGGELING L, SAHM H. Functional and structural analyses of threonine dehydratase from Corynebacterium glutamicum [J]. Bacteriol, 1992, 174: 8065-8072.
[24] SHARMA R, KESHARI D, SINGH K S, YADAV S, SINGH S K. MRA_1571 is required for isoleucine biosynthesis and improves mycobacterium tuberculosis H37Ra survival under stress [J]. Scientific Reports, 2016, 6: 27997.
[25] DONG Xun-yan, ZHAO Yue, HU Jin-yu, WANG Xiao-yuan, LI Ye. Attenuating l -lysine production by deletion of ddh and lysE and their effect on L-threonine and L-isoleucine production in Corynebacterium glutamicum [J]. Enzyme and Microbial Technology, 2016, 93/94: 70-78.
(Edited by YANG Hua)
Foundation item: Project(31260476) supported by the National Natural Science Foundation of China
Received date: 2016-09-25; Accepted date: 2016-10-30
Corresponding author: LIANG Zhi-huai, Professor, PhD; E-mail: Liangzhihuainky@163.com; WU Cai-jun, Professor, PhD; E-mail: wucj12@126.com
Abstract: In order to screen the genes controlling watermelon rind color and luster, the experiment was carried out with yellow watermelon skin mutants as tester and green wild type watermelon as control, and transcriptome sequencing and bioinformatics analysis were done. The results show that 34.27Gb clean data were got by transcriptome sequencing. There are 261 differentially expressed genes among Y1_vs_G1, Y2_vs_G2 and Y3_vs_G3. The pathways contenting most differentially expressed genes are plant hormone signal transduction pathway, phenylpropanoid biosynthesis pathway, photosynthesis pathway, starch and sucrose metabolism pathway. 9-cis-epoxycarotenoid dioxygenase (Cla002942), alcohol dehydrogenase (Cla004992), photosystem I reaction center subunit III, chloroplastic (precursor) (Cla009181), long-chain acyl coenzyme A synthetase (Cla017341), threonine dehydratase biosynthetic (Cla018352) candidates genes were screened out.
