

Microbial community structure and function in sulfide ore bioleaching systems
QIU Guan-zhou(�����), LIU Xue-duan(��ѧ��), ZHOU Hong-bo(�ܺ鲨)
Key Laboratory of Biometallurgy of Ministry of Education,
School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China
Received 20 September 2008; accepted 5 November 2008
Abstract:
The severe current situation facing to minerals processing is that the most minerals are characterized by low-grade, being complex and very hard to deal with. It is necessary to find a new way to solve these questions. Nowadays, biohydrometallurgy draw more and more attention because of its simple process, low cost and kindness to environment. However, the lack of suitable bacteria and hard research on the mechanisms between the bacteria and ores or bacteria in gene level result in the low efficiency and poor yield of the target metal in bioleaching. Therefore, the understanding of the microbial community structure and function in the bioleaching systems is very important for the optimization of microbial community by controlling the operating conditions in bioleaching systems, thus enhancing the leaching rate. A review is given on the achievements and progress related to the study on microbial community structure and function in sulfide ore bioleaching systems made in our research group.
Key words:
bioleaching; biohydrometallurgy; community structure; community function;
1 Introduction
The development of biohydrometallurgy will make it possible to enlarge the amount of mineral deposits that can be exploited, and guarantee the mineral resources demanded for the fast economic development. The basic research on bioleaching of low-grade and complex sulfide ores is of important significance for the treatment of the minerals in China[1].
The bioleaching of low-grade and complex sulfide ores is characterized with low leaching rate and low metal extraction. The microorganisms involved in the bioleaching process are mainly acidophilic chemolitho- trophic species, which grow slowly and result in low extraction of metals. Otherwise, the interactions of different groups of bioleaching microorganisms and relationships between microorganisms and environment also influence the dissolution of sulfide ores[2-3]. Therefore, understanding the microbial community structure and function in the bioleaching systems, as well as the relationships of the population dynamics with the environmental factors is very important for the optimization of microbial community by controlling the operating conditions in bioleaching systems, thus enhancing the leaching rate[4]. To solve this problem, the project ��The Basic Research of Microbial Hydro- metallurgy�� funded by Ministry of Science and Technology of China and the project ��The Basic Research of Bioleaching of Sulfide Minerals�� supported by the National Natural Science Foundation of China focus on the investigation of ecological succession of micro- organisms during sulphide-bioleaching, the screening of bioleaching microorganisms and the complex interfacial interactions among microbe, mineral and solution. In this work, the achievements and progress related to the study on microbial community structure and function in sulfide ore bioleaching systems are reported.
2 Achievements and progress
2.1 Basic research of bioleaching microorganisms
2.1.1 Development of a novel strategy for rapid and accurate screening of highly-efficient Acidithiobacillus ferrooxidans in various environments by the ��Standard for Identification of Acidithiobacillus ferrooxidans and Their Activity��Acidithiobacillus ferrooxidans is one of the most important and widely used bacteria in bioleaching process. Through cooperation with TIGR (USA), we managed to obtain the sequence information of 3 217 genes of the model bacteria Acidithiobacillus ferrooxidance ATCC 23270[5] (shown in Fig.1) as early as July 2004, which was firstly reported in China. Then specific and representative probes (58-mer in length) were developed for each of the 3 217 genes using a Genomic Array Probe Designing Programme. The probes were synthesized and the whole genomic array of Acidithiobacillus ferrooxidans ATCC 23270 was fabricated. Ten human genes and ten genes from plants were planted as negative control.
Fig.1 Whole genomic schemtic of A.f ATCC 23270
Based on flask shaking, column soakage and pile soakage experiments conducted in different mining areas in China, we acquired a series of Acidithiobacillus ferrooxidans bacteria with various oxidation capacities. Then the DNA of these bacteria was extracted and hybridization experiments were conducted using the DNA and the whole genomic array of bacteria ATCC 23270. The results show that among the 3 217 genes, 967 genes are specific in different strains of bacteria, and the remaining ![]() genes commonly exist in Acidithiobacillus ferrooxidans (as shown in Fig.2). We have discovered 320 characteristic genes related to high oxidation capacity. Among these genes, 24 are related to S and N metabolism; 33 are related to the biosynthesis and degradation of murein, peptidoglycan and lipopolysaccharide; 15 are related to detoxification processes; 36 are related to DNA metabolism; 85 are related to energy metabolism; 5 are related to the metabolism of fatty acid and phosphoric acid; 26 are related to protein synthesis; 12 are related to nucleotide synthesis; 15 are related to transcription; 34 are related to function regulation; 10 are related to information transformation; and 25 are related to cellular transportation and binding protein. Moreover, 135 genes related to the oxidation of Fe2+ and S as well as the stress resistance are also discovered. A comprehensive utilization of these discoveries leads to a new standard to evaluate the biohydrometallurgical ability of the Acidithiobacillus ferrooxidans bacteria of interest. The ��National Standard for the Identification and Determina- tion of the Activity of Acidithiobacillus ferrooxidans using Gene Array�� was approved as a national standard in 2007[6].
genes commonly exist in Acidithiobacillus ferrooxidans (as shown in Fig.2). We have discovered 320 characteristic genes related to high oxidation capacity. Among these genes, 24 are related to S and N metabolism; 33 are related to the biosynthesis and degradation of murein, peptidoglycan and lipopolysaccharide; 15 are related to detoxification processes; 36 are related to DNA metabolism; 85 are related to energy metabolism; 5 are related to the metabolism of fatty acid and phosphoric acid; 26 are related to protein synthesis; 12 are related to nucleotide synthesis; 15 are related to transcription; 34 are related to function regulation; 10 are related to information transformation; and 25 are related to cellular transportation and binding protein. Moreover, 135 genes related to the oxidation of Fe2+ and S as well as the stress resistance are also discovered. A comprehensive utilization of these discoveries leads to a new standard to evaluate the biohydrometallurgical ability of the Acidithiobacillus ferrooxidans bacteria of interest. The ��National Standard for the Identification and Determina- tion of the Activity of Acidithiobacillus ferrooxidans using Gene Array�� was approved as a national standard in 2007[6].
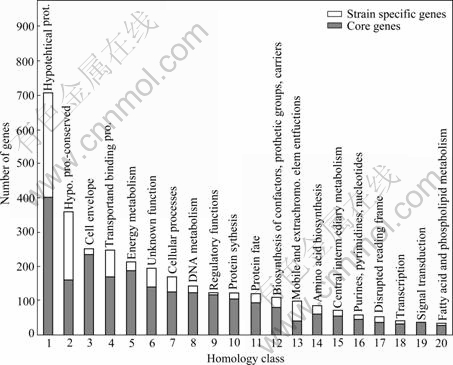
Fig.2 Distribution of common and specific genes in different A.f
Based on the whole genomic array of the model bacterium ATCC 23270, we can identify the capability of the bacteria at the gene level. This strategy has shortened the period of identification of bioleaching bacteria from several months to 3-5 days. Furthermore, this genomic- arrayed-based strategy assesses the capability of the bacteria directly from the related genes and hence the result is environmentally proven in contrast to traditional methods[7-8]. This standard provides a simple, quick and accurate method for the screening of Acidithiobacillus ferrooxidans bacteria from different environment and thus is of significance in solving the problem of low efficiency in the biohydrometallurgical process.
2.1.2 Development of microarrays and technology systems to study bio-mining microbial community and function
1) Establishing biomining community genome microarray (CGA)
The community genome microarray is shaped by directly putting the genome DNA from the pure cultured bacteria on the slides[9-10]. The microarray was developed by the members of the work group in the Oak Ridge National Laboratory, USA. To date, the bio-mining community genome microarray contained most of bio-mining microorganisms affiliated with 15 species (or more than 50 strains). The elementary results showed that the microarray under hybridization conditions of 55 �� temptations and 50% foraminde has better interspecies speciality, better sensitivity (up to 0.1 ng genome DNA) (Fig.3) and better quantitative capability.

Fig.3 Hybridization amount of genome DNA with community genome microarray
2) Establishing bio-mining function genome micro- array (FGA)
The FGA is established by using the functional genes related to metabolic activity as probes[11-12]. The BM FGA, used for the bio-mining microbial community and functional analysis, included not only the functional genes but also 16S rRNA genes that reflected the microbial phylogenetic development and taxonomic status. This array contained 1 072 probes in which there were 571 related to 16S rRNA and 501 related to functional genes. The functional genes in the microarray were involved in carbon metabolism (158), nitrogen metabolism (72), sulfur metabolism (39), iron metabolism (68), DNA replication and repair (97), metal-resistance (27), membrane-relate gene (16), transposon (13) and IST sequence (11). Almost all of the probes are affiliated with 35 species of acidophilic microorganisms and 9 species of related subclass, in addition of some iron mountain clones, detected in the natural acid environment and bioleaching systems. Based on the results of microarray hybridizations, specificity tests with representative pure cultures indicated that the designed probes on the arrays appeared to be specific to their corresponding target genes. The detection limit was 5 ng of genomic DNA in the absence of background DNA. Moreover, strong linear relationships between the signal intensity and the target DNA were observed (r2��0.98).
3) Developing microarray-based approach for whole community genome amplification to detect lower nanogram of community genome DNA and greatly improve the sensitivity of microarray for detection of microbial community
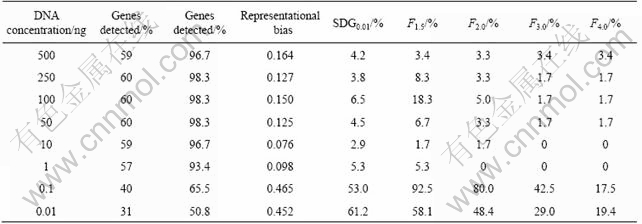
A problem in the application of microarray is that its sensitivity is low for the analysis of microbial community in environments. Through cooperation with Oak Ridge National Laboratory, we developed an approach for whole community genome amplification. The approach can amplify genomic DNA lower than 10 fg (Fig.4). When the approach was applied to the environment samples, the results showed that most equivalent genes (93%-97%) were detected when the biomass of the module DNA was over 1 ng (Table 1). Here, the detection of microarray for microbial community was representative.
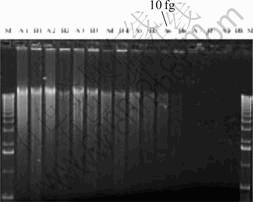
Fig.4 Electrophoresis with different amounts of module DNA
Table 1 Representative detection of genes from environmental sample
The above approach and system were first applied to the analysis of bio-mining microbial communities and the analysis of the relationship between the structure and function of the microbial community in bio-mining environments[13]. Combining the environmental parameter with mineral elements in bio-mining systems, we can forecast and control the microbial community, so as to optimize the industrial procedures and improve the effect of bio-mining.
2.2 Microbial community structure and function
2.2.1 Microbial community structure analysis of acid mine drainage
1) Firstly the relationship between the diversity of microbial communities in Chinese biomining environments and biogeochemical characteristic was clarified based on the molecular cloned techniques.
Thirty-two water samples from the acid mine drainage and the mined liquid pond were collected from thirteen mine districts in China (Fig.5), and the biogeochemical characteristic such as pH and twenty-one chemical elements were examined. The results showed that the biogeochemical characteristics from the Guangxi Dachang, Liuyang Qibaoshan and Gansu Baiyin mine districts were distinctly different from the others.
Fig.5 Location of sampling stations for analysis on diversity of microbial communities
Based on the molecular markers such as the 16S rRNA, GyrB and iro genes and the PCR-RFLP analysis, we recovered a total of 275 16S rRNA, 130 GyrB and 56 iro sequences from 32 sample sites (13 mine districts). These results provided a large resource of gene sequences for later miroarray studies. Internationally, we were first to use the GyrB gene to study the distribution of microbial communities in biomining environments and found that the GyrB gene could more exactly reflect the essence of microbial community than the 16S rRNA gene. In these studies, we also found that the GyrB gene from the biomining microorganisms was distinctly different from those in the GenBank databaseThe related phylogenetic trees showed that L.f, A.ferrooxidans and a possible new microorganism were dominant groups in the Chinese acid mine drainage. Moreover, the RFLP and 16S rRNA sequences analysis showed that the heterotrophic acidophilic microorganism (Acidiphilium) was extensively detected but it was not a dominant group. Likewise, the predominant groups had great differences in different sites. For instance, A.f was the dominant group in ZJ site but L.f was detected in DWT and SLS. 16S rRNA sequences analysis also showed that a kind of microorganism possibly belonging to a new genus or species was extensively found in the mine districts and had a rather high proportion in the total microbial community, especially in the ZJ site from the Dexing Copper Mine, Jiangxi Province, China. The microorganism had 40% proportion.
Through the combined analysis of the microbial community structure and the biogeochemical characteristics[14], we found that the mine districts with the different biogeochemical characteristics had different microbial community structures, for example, Gansu Baiyin mine districts and Jiangxi Dexing Copper Mine. Furthermore, even if the sites were located at the same mine district, they possibly had different microbial community structures, for example, the KZX site and DWT site, which were both located at the Jiangxi Dexing Copper Mine. By the combined results from all sites, we concluded that pH has an important influence on the distribution of L.f. Under conditions of low pH value, L.f was the dominant group in such sites as the Guangxi Dachang Gaofeng 1 (pH 1.7), Jiangxi Dexing DWT and SLS (pH 2.0). In addition, redox potential and the toxic metal iron also have important effects on the microbial communities.
2) Analysis of microbial community structure and function in acidic environmental samples by applying 50-mer oligonucleotide arrays
When the 50-mer oligonucleotide arrays were used as a generic profiling tool to reveal differences among various microbial communities, bulk community DNAs from the natural acidic bio-niche (NAE) (AMD sample) and the bioleaching system(BLS) were acquired from the acidic water (NAE was the natural bio-niche with pH 2.0 and BLS was the bioleaching system with pH 2.5). For comparison, DNA from a weak acidic water sample, CWAE with higher pH 5.0, was used as a reference. 5 mg of the purified bulk community DNA from three samples was directly labeled with Cy3 using random-primer labeling. The Cy3-labeled DNAs were hybridized with the microarrays. All spots with SNRs of ��2 were considered as positive signals. Overall, the numbers of 16S rRNA probes with statistically significant positive signals were 80 (37 in the BLS sample, 42 in the NAE sample and 23 in the CWAE sample). These 16S rRNA probes were mainly related to 15 different genus or species, including Acidithiobacillus ferrooxidans, Leptospirillum sp., Acidithiobacillus thiooxidans, Acidiphilum spp., Sulfobacillus sp., Holophaga sp., Metallosphaera sp., Alicyclobacillus herbarius, Ferroplasma spp., Thiomonas sp., Alicyclobacillus spp., Alternaria tenuissima, Thermoplasmataceae archaeon, Desulfosarcina variabilis and Acidianus sp. However, the composition and richness determined by microarray- based analysis of microbial community were different in all three sites. For example, Acidithiobacillus ferrooxidans, Leptospirillum sp. and sulfobacillus sp. were dominant in NAE and BLS samples and the signal intensity level was far higher than that of the CWAE sample. However, there was a distinct heterogeneity for these dominant genus or species at NAE and BLS samples.
When the 50-mer oligonucleotide arrays were used as a generic profiling tool to reveal functional differences among various microbial communities, a total of 150 functional gene probes mainly involved in nitrogen metabolism, carbon metabolism, sulfur metabolism, iron metabolism and metal-resistance produced significant positive signals (SNR��2) in all three samples. The numbers of detected functional gene probes were 35, 118 and 116 in CWAE, NAE and BLS samples, respectively. The microarray analysis of the functional genes indicated that NAE and BLS samples were distinctly different from CWAE sample. NAE and BLS samples resulted in higher signal intensity of functional genes related to carbon metabolism, nitrogen metabolism, iron metabolism and metal-resistance. However, the hybridization signal intensity of these functional genes was significantly reduced when the extract DNA of the CWAE was used in the hybridization process[15].
The cluster analysis based on 16S rRNA showed that the BLS sample was more closely clustered with the NAE sample than the CWAE sample, suggesting that the microbial community structure in the BLS sample was more similar to that in the NAE sample than that in the CWAE sample. Moreover, functional genes detected in the BLS sample also more closely clustered with the NAE sample than with the CWAE sample, suggesting that the community structure in terms of components of the functional genes was also most similar to that of the CWAE sample.
Importantly, the general trend based on the PCA analysis of geochemical characteristics and cluster analysis showed that the sample with the similar environmental variables had the similar microbial community composition and functional gene distribution.
3 Outlook
Aiming at bio-extraction of sulfate mineral and on the basis of the researches about the genetic polymorphism, species diversification and functional genome information, we studied the community structure of the microbial of interest and the fluctuation of their composition and structure and managed to find a brand-new strategy for the screening of highly efficient bioleaching microorganisms. All these works provide a strong basis for later research works. We plan to focus researches on the following aspects:
1) Rapid screening of bioleaching bacteria particularly for soakage of raw sulfate minerals
Based on the screening system built up, including study on species diversification, important functional genes and molecular polymorphism in the bacteria, we will be able to focus on the screening of highly efficient bacteria and carry out research on the rapid screening strategy and functional genome research on bioleaching bacteria.
Using A.f whole genomic array, community genome array and functional gene array, we will isolate strains of highly efficient bioleaching bacteria, design and optimize consortia for various mineral and environment. Meanwhile, we will continue the isolation of thermophilic bacteria in western China in order to find some bacteria that are adapted to the peculiar climate and environment and are especially efficient in soakage of western mineral recourses.
2) Functional genomic study on bacteria highly active or resistant to high temperature
We will study the changes of gene expression spectrum of the bacteria under stresses such as pH, temperature, ionic heavy metal and organic compounds in order to explain the corresponding response mechanisms directly related to mineral soakage in the gene level. Through gene prokaryotic expression, mutation experiments, gene array analysis and protein spectrum analysis, we will be able to discover more functional genes and elucidate the regulatory networks. Besides, we will conduct the whole genomic sequencing of L.f in cooperation with Lawrence Berkeley National Laboratory, USA. This will be the first time for the whole genomic sequencing work for L.f bacteria.
3) Study on community structure of bioleaching microbial and interaction among different microbial communities
With the application of the gene arrays already built up, we might be able to find out the functional and structural differences in bioleaching microbial communities and the overall alternation of these communities and their inner structure. Combining with analysis of mineral components and environmental parameters of the bioleaching systems, we should be able to elucidate the regulatory mechanisms for changes in community structure, microbial activity and interaction among different microbial communities. For bacteria already isolated, we will conduct researches on the molecular polymorphism and their evolution. We will also conduct functional molecular labeling experiments to study the genetic polymorphism for bacteria in different mining areas. The newly cloned genes and genomic information of successively isolated bacteria will be added to the array already built, forming the second generation of functional gene array and community genome array. These will help a lot in research on the community structure and the functions of various bioleaching bacteria.
4 Conclusions
More than 70% of non-ferrous metal mineral resources in China are low-grade and complex sulfide ores. Difficulty of hydrometallurgy of such ores lies in the lack of specific, adaptive environmentally-friendly bacteria, causing slow leaching and low extraction rate. Method of a simple, rapid and accurate biological metallurgy rapid screening of bacteria gene chip created from the level of genes, not only makes bacteria leaching test shorten from a few months to 3-5 days, but also overcomes shortcomings of the traditional method of leaching, such as the determination of only phenotypic characteristics, being affected greatly by environment and the instability of the results. This is due to the evaluation of its performance through biological control of the gene chip leaching metallurgical microbial genetic traits related to the comprehensive testing. The realization of building a chip on the leaching process of microbial population changes in the quantitative analysis, genes analysis of the microbial community structure and function combined with macro-engineering, physical and chemical factors in the hydrometallurgy process, and optimized control of the structure of microbial community, make hydrometallurgy from the traditional descriptive study to the current-forecast across studies. The use of the screening of bacteria for hydrometallurgy has greatly reduced the quality of the border of mineral resources, and expands the storage of non-ferrous metals, which has a good prospect of application.
References
[1] ZHOU J K, NIU Y J. Advance in research of biological metallurgy of sulfide ore [J]. Metal Mine, 2005, 346(4): 24-30.
[2] GALLEGUILLOSA P, REMONSELLEZA F, GALLEGUILLOSA F, GUILIANI N, CASTILLO D, DEMERGASSO C. Identification of differentially expressed genes in an industrial bioleaching heap processing low-grade copper sulphide ore elucidated by RNA arbitrarily primed polymerase chain reaction [J]. Hydrometallurgy, 2008, 94(1/4): 148-154.
[3] ZENG Le-ping,HUANG Ju-fang, ZHANG Yan-fei, QIU Guan-zhou, TONG Jian-bin, CHEN Dan, ZHOU Jin, LUO Xue-gang. An effective method of DNA extraction for bioleaching bacteria from acid mine drainage [J]. Applied Microbiology and Biotechnology, 2008, 79(5): 881-888.
[4] ZHANG Cheng-gui, XIA Jin-lan, WANG Jing, QIU Guan-zhou. Progress on researches of sulfur oxidation system of Acidithiobacillus spp [J]. Biotechnology Bulletin, 2007, 24(1): 59-65.
[5] QIU G Z, LIU X D, LIU J S, WANG J. The methods of testing Acidithiobacillus ferrooxidans and its activity by microarray technology [S]. 2006.
[6] YIN Hua-qun, CAO Lin-hui, QIU Guan-zhou, WANG Dian-zuo, KELLOGG L, ZHOU J Z, DAI Zhi-min, LIU Xue-duan. Development and evaluation of 50-mer oligonucleotide arrays for detecting microbial populations in Acid Mine Drainages and bioleaching systems [J]. Journal of Microbiological Method, 2007, 70: 165-178.
[7] YIN H Q, QIU G Z, WANG D Z. Comparison of microbial communities in three different mine drainages and their bioleaching efficiencies to low grade of chalcopyrite [J]. Journal of Central South University Technology, 2007, 14: 460[7]466.
[8] GARRIDO P, GONZ?LEZ-TORIL E, GARC?A-MOYANO A, MORENO-PAZ M, AMILS R, PARRO V. An oligonucleotide prokaryotic acidophile microarray: Its validation and its use to monitor seasonal variations in extreme acidic environments with total environmental RNA [J]. Environmental Microbiology, 2008, 10(4): 836-850.
[9] ZHOU J Z. Microarrays for bacterial detection and microbial community analysis [J]. Current Opinion in Microbiology, 2003, 6(3): 288-294.
[10] KRAUSE D O, SMITH W J M, McSWEENEY C S. Use of community genome arrays (CGAs) to assess the effects of Acacia angustissima on rumen ecology [J]. Microbiology, 2004, 150: 2899-2909.
[11] ZHANG Yu-guang, LI Di-qiang, XIAO Qi-ming, LIU Xue-duan. Microarrays and their application to environmental microorganisms [J]. Acta Microbiologica Sinica, 2004, 44(3): 406-410.
[12] MAERCKER C. Protein arrays in functional genome research [J]. Bioscience Reports, 2005, 25(1/2): 57-70.
[13] WU Li-you, LIU Xue-duan, SCHADT C W, ZHOU J Z. Microarray- based analysis of subnanogram quantities of microbial community DNAs using whole community genome amplification (WCGA) [J]. Applied and Environmental Microbiology, 2006, 72(8): 4931-4941.
[14] ZHANG Y G, ZHANG X Q, LIU X D. Microarray-based analysis of changes in diversity of microbial genes involved in organic carbon decomposition following land use/cover changes [J]. FEMS Microbiology Letters, 2007, 266(2): 141-155.
[15] YIN Hua-qun, QIU Guan-zhou, LUO Hai-lang, CAO Lin-hui, DAI Zhi-min, WANG Jie-wei, WANG Dian-zuo, LIU Xue-duan. The research of ferrous iron-oxidizing capability of Acidithiobacillus ferrooxidans and the efficiency of low grade chalcopyrite bioleaching [J]. Progress in Modern Biomedicine, 2007, 7(5): 641-642.
Foundation item: Project(50621063) supported by the National Natural Science Foundation of China; Project(2004CB619200) supported by the National Basic Research Program of China
Corresponding author: QIU Guan-zhou; Tel: +86-731-8877212; E-mail: qgz@mail.csu.edu.cn
(Edited by YANG Bing)
