
������ȼ�ջ���β��������Ca(OH)2�Ĺ�������
���ڣ����ػ�����ƽ��������������
(����������ѧ ������ѧ�빤��ѧԺ������ ������650500)
ժ Ҫ��
ȼ�ջ���β������ȼ�չ����м����������ƣ��Ի���β��ȼ�ղ�����������������ȥ�����ı��ȼ�ȿ��췴Ӧ�¶ȶ�ȥ���ʵ�Ӱ�죬�����ռ�����XRD��SEM��EDS�����������Ԫ�ط���������FactSage6.2����ѧ���������ɵĹ���������Ԥ�⡣�о����������������Ԥ��ȼ�ջ���β��������¶�1 060 �棬�ڿ�ȼ��1.7~4.2�ķ�Χ��¯���¶����ȶ���870 �����ϡ����ռ���ͬ����������Ӧ�����������¶����߶����ӣ�1 060 ��ʱ������Ϊ86%��920 ��ʱ������Ϊ50%��870 ��ʱ������Ϊ30%����������ͬ���������������γɼ�ʽ����ƣ�֮���Ϊ����ƣ��ڸ����������ת��Ϊ������ơ���������Ԥ������ʵ����һ�¡�
�ؼ��ʣ�
��ͼ����ţ�X511 ���ױ�־�룺A ���±�ţ�1672-7207(2013)02-0835-08
Phosphorus-fixation by hydrated lime in fluidized bed combustion of yellow phosphorus tail gas
WANG Lei, WANG Zhonghua, NING Ping, JIANG Ming, QIN Yangsong
(Faculty of Environment Science and Engineering, Kunming University of Science and Technology, Kunming 650500, China)
Abstract: Hydrated lime was tested for removing phosphoric pentoxide during the premixed combustion of yellow phosphorus tail gas in fluidized bed. The effect of temperature on the retention of phosphoric pentoxide in hydrated lime was examined by changing air fuel ratio. XRD, SEM and EDS techniques were used to analyze and characterize fresh sorbent and sorbent after reaction. The computer software package FactSage6.2 was used to predict solid product. The results show that the highest temperature of yellow phosphorus tail gas premixed combustion in fluidized bed is 1 060 ��. Within flow ratio of air to fuel from 1.7 to 4.2, the temperature can be stabilized above 870 ��. The sorbent can react with phosphoric pentoxide and removal efficiencies increase with the increase of the temperature. Phosphoric pentoxide removal by hydrated lime is 86% at 1 060 ��, 47% at 920 ��, and 30% at 870 �� in fluidized bed, respectively. The sorbent reacts with phosphoric pentoxide firstly forming basic calcium phosphate, and then changing into calcium phosphate. At high temperature, calcium phosphate changes into calcium pyrophosphate. Solid product predicted results are consistent with the experimental results.
Key words: phosphorus-fixation; premixed combustion; yellow phosphorus tail gas
�����ǹ��õĻ���ԭ���ϣ��ڹ����о��й㷺��;��ÿ����1 t���ף���������β��2 500~3 000 m3��β���к�CO��������Ϊ87%~92%����������Ũ��Ϊ0.8~8.0 g/m3����������Ũ��Ϊ0.5~1.5 g/m3������һЩ���ʣ�����ֵ10.5~11 MJ/m3[1-2]�����кܸߵ����ü�ֵ���й��Dz��״��������ռ�����ܲ��ܵ�80%��2010����ײ���89.9��t��������ŷ�Լ24��m3�Ļ���β��[3]��Ŀǰ���ڻ���β�������ʲ���30%��������ҵ����ȼ�պ�ֱ���ŷ�[4]�������������Դ�˷ѣ��Դ�������Ҳ�������Ⱦ��Ŀǰ�ۺ����û���β������Ҫ;��Ϊ������Դ����Ϊһ̼����ԭ���Լ�����β������[5-6]��������β��������Դ���ԭ�ϣ��������ޣ�ȼ�պ����岻�������ŷţ���ɻ�����Ⱦ������̼һ��ѧƷ�ϳɣ���Ȼ����˻���β�������ü�ֵ��������β���ɷָ��ӣ������豸Ͷ�ʴ�����β�����磬��������¯ȼ�ջ���β�������У�����ḯʴ¯���ڻ������ͻ��ȹܱ�[7-9]��7~90 d֮�ھ��ܵ��´���[10]����¯ͣ�á��ڴˣ��������߲���һ������������ȼ�յ���������Ԥ��ȼ�ջ���β������ȼ�չ����м����������ƶ���[11]���������գ������������ƵĹ������á���Ŀ������̽��һ�ּȲ���ʴ�豸�����ܽ��ͻ�����Ⱦ��ͬʱ���ܸ�Ч���û���β�����ܵķ�����
1 �ֳ�ʵ�鼰�������
1.1 ʵ��ԭ��
1.1.1 β������Դ�����
����β����Դ��ij���׳�����¯�����������Ļ���¯������3��ˮ��������������پ�3��ˮϴ���õ�β����������λ�ھ���װ�����ǰ��������ձó�ȡ������β����Ҫ������1��ʾ������������Դ�ڸû��׳������ֵ��PH3��H2SΪ��ʵ��������ֳ��������
��1 ����β����ɳɷ�
Table 1 Components of yellow phosphorus tail gas
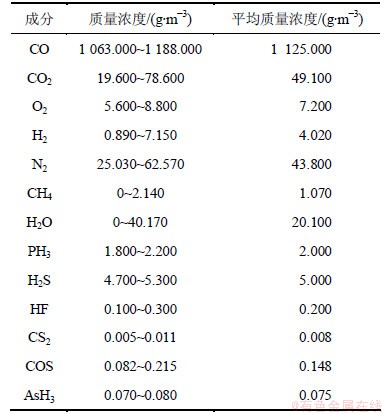
1.1.2 ���ռ����Ʊ�
ѡ���Լ�Ϊ������Ca(OH)2 (�ɶ��п��������Լ���)��Ca(OH)2����������ˮ��ϣ�����������飬��ɸ�Ƴ�ֱ��Ϊ1.0~1.2 mm�Ŀ�����
1.2 ʵ��װ�ú���
1.2.1 ����С��������װ��
ʵ����ͼ1��ʾ��С����������Ӧװ���н��С����й���ϵͳ����һ������Ϊ3.5 m3/h����ձ÷����������β����һ������Ϊ7.0 m3/h����ձ÷������������ʹ���������ڷ���ת�������ơ����ػ�����U��ѹ���ƽ��в����Ϳ��ơ�ȼ��ϵͳΪ����С������������Ԥ���ҡ�ȼ�����졢����塢¯��(�ھ�56 mm���߶�600 mm)��ɡ����ⲿ�ù�������ά�������£�¯���¶ȵIJ�����2��S�Ͳ����ȵ�ż�ֱ����Ӳ�������ɡ��ȵ�ż���·�ΧΪ0~1 300 �棬�����ҵľ���ֱ�Ϊ70 mm��500 mm��ȼ�պ����徭����ˮ����ȴ�ܺͲ����������������ŷš�
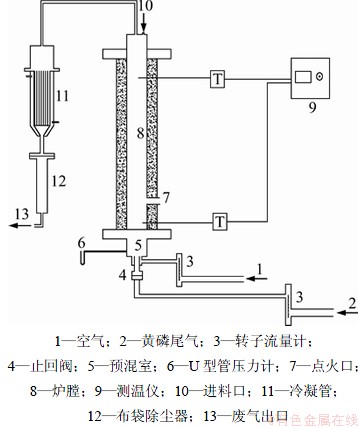
ͼ1 ������ʵ��װ��ʾ��ͼ
Fig.1 Schematic diagram of experimental setup
1.2.2 ��Ӧ���̼������
������¯���ڼ���100 g���ռ���Ȼ��ͨ������ͻ���β������������Ԥ�����ڻ�ϣ������ͨ����������¯������ȼ��ͬ���ռ���Ӧ������ʵ�������ͨ��U��ѹ���Ʋ������ѹ����1~2 kPa��Χ�ڡ�ʹ���������ڷ�������β������������1 m3/h���ң�ͨ���ı����������ò�ͬ�Ŀ�ȼ��(���������������β������֮��)���ڲ�ͬ�Ŀ�ȼ���£��ⶨȼ���¶ȣ��¶��ȶ�5 min���¼����ֵ����Ӧ��ɺرտ�����ȼ�����ã������ռ���¯������Ȼ��ȴ��ȡ����
1.2.3 ��Ӧǰ��������
��ʵ��װ��ǰ����������ܲ���������Ļ���β����PH3��H2S�ĺ�����ÿ��ʵ��ǰ�ֱ��������һ�Ρ�����PH3��������Ϊ0.1~1.5��0.2~2.5 g/m3��H2S��������Ϊ0.2~5.0��2.0~10.0 g/m3������PH3ȼ�պ������P2O5��300 �����±�Ϊ�����������������ˮ������������ļ�ⲻ�ܷ�Ӧ���ռ���ʵ��ȥ���ʡ����о��У�ͨ���ⶨ��Ӧ��������ռ��е�����������Ca(OH)2�Ĺ���Ч����
1.3 ���ռ��������
(1) ���ռ��������IJⶨ�����������ֹ��ȷ�(GB 4701.7��85)���ⶨ�����п���Ca���ӵ�Ӱ��ʹ������������������ȡ�
(2) ���ռ�����ṹ����ʹ���ձ���ѧTTRIII��X��ת��������(XRD)������Cu�У��ܵ�ѹ40 kV���ܵ���200 mA��ɨ������10 (��)/min��ɨ�跶Χ5��~90�㣻�ձ���ѧD/Max-3B��X��������(XRD)������Co�У��ܵ�ѹ20 kV���ܵ���20 mA��ɨ������10 (��)/min��ɨ�跶Χ13��~110�㡣
(3) ���ռ�������ò����ʹ�÷�����XL30ESEM-TMP�ͻ���ɨ��羵(ESEM)�����Ե�ѹ20 kV����Ԫ�ط���ʹ�������˹Phoenix-OIM��X��������(EDS)��
1.4 ȥ��Ч�ʼ���
��ȥ��������100 g���ռ�����������������ȼ�չ����л���β�������������������õ��������������β����PH3����Ũ�Ȼ����ȶ���2 g/m3����, ����������1 m3/h, β���������������ȡ����ȼ��ʱ��(��������β��ʱ��)�����ռ��к�����������ͬһ��Ʒ2�βⶨ��ƽ��ֵ����ó������ռ�������β������������P2O5�ļ�����ʾ��
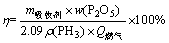 (1)
(1)
ʽ�У���Ϊ��ȥ��Ч�ʣ���(PH3)ΪPH3��������Ũ�ȣ�m���ռ�Ϊ���ռ���������w(P2O5)Ϊ���ռ��е�������%��Qȼ��Ϊ����β����������2.09ΪPH3��P2O5��ת��ϵ����
2 ����ѧ����
Ϊ�����ʵ�������Լ���֤ʵ�����Ŀɿ��ԣ���ʵ����е�ͬʱ����FactSage6.2[12-13]����ѧ���������ķ�Ӧģ�顰Reaction������Ԫ����ƽ��ģ�顰Equilib���� ���������ݿ�Fact53�����������м����Ԥ�⣺
(1) ��ͬ��ȼ���»���β���ľ���ȼ���¶ȣ�
(2) ���¶ȷֱ�Ϊ920��1 060 �棬ѹ��Ϊ101.3 kPa�����£����ݼ���˹��������С��ԭ�����Է�Ӧ�����ռ��еĹ���������Ԥ�⡣
3 ���������
3.1 ��ȼ�ȶ�ȼ���¶ȵ�Ӱ��
��1.2.2������������������Ԥ��ȼ�ջ���β������ͬ��ȼ����ʵ���¶���ͼ2��ʾ��
Ϊʹʵ����������ѧ������֮����ڿɱ��ԣ�����ѧ���������������Ҫ����2�����أ�
(1) �ɱ�1��֪��CO��CO2�ڻ���β����ռ���֣�����������¶�Ϊ25 �棬ѹ��Ϊ101.3 kPaʱԼΪ1 m3, ��ˣ�����β����ΪCO��CO2 2����֣�CO��������Ϊ1 125 g, CO2��������Ϊ49 g��
(2) �������¶Ⱥ�ѹ���£�1 m3������O2 248 g��N2 932 g���Դ�Ϊ�����ı����������ʹ��ȼ����1.5������4.5��ʹ�÷�Ӧģ�顰Reaction�� �Ի���β������ȼ���¶�(��H=0)�����˼��㣬�����ͼ3��ʾ��
��ͼ2��ͼ3���Կ�������ȼ�ȶ�ȼ���¶��кܴ��Ӱ�죬�¶����ȼ�ȵı仯���ƻ���һ�£�����ȼ��Ϊ2.5����ʱȼ���¶ȶ�����һ����ߵ㡣�¶���ߵ㸽��ȼ���Ϳ�����ȫ��Ӧ���������������㣬ȼ����ȼ�շ�Ӧ����ȫ��ȼ���¶�������������Ӷ����ӣ����Ҳ������������Ȼȼ��ȼ�ճ�֣��������������ڼ��ȹ���������ȼ���¶�����������������Ӷ����͡����ھ��ȼ���û�п���������ʧ��ʵ��ȼ���¶ȱ�Ȼ�������ۼ���ľ���ȼ���¶ȡ�
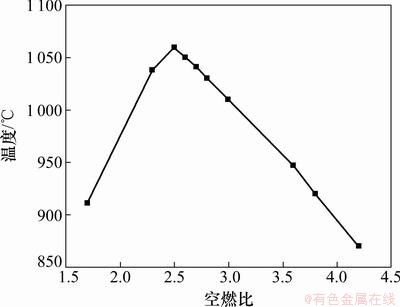
ͼ2 ��ͬ��ȼ��ʱȼ���¶�����
Fig.2 Premixed combustion temperature curve at different flow ratios of air to flue
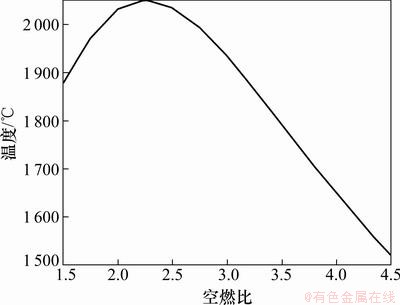
ͼ3 ��ͬ��ȼ��ʱ����ȼ���¶�����
Fig.3 Adiabatic combustion temperature curve at different flow ratios of air to flue
ͨ���Գ�������ļ�⣬ֻ�ڿ�ȼ��Ϊ1.7��2.3ʱ����H2S��PH3��Ҳ֤ʵ���������ۡ���ˣ�����ʵ��ѡ���ȼ��Ϊ2.5��3.8��4.2�����½��С�
3.2 ȼ������ȥ����
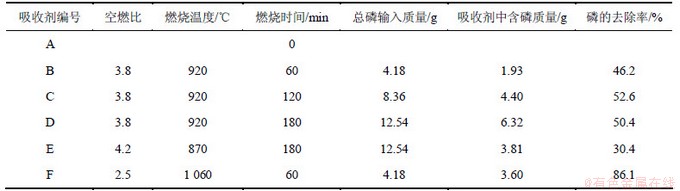
��ȼ�շ�Ӧ������ռ�����ǰ����������������������ռ��е�����������ʽ(1)�������ȥ���ʣ����ý������2��
�ӱ�2���Կ��������ռ��Ի���β��ȼ�պ������е�����ȥ�����ã��ڷ�Ӧ�¶�920 ��ʱ�����ռ��е������淴Ӧʱ����ӳ������ߣ����淴Ӧʱ����ӳ�β��������������ӣ�ȥ��Ч�ʲ���ʱ��ɱ��У�����Ӧʱ����ͬʱ(���ռ�B��F�������ռ�D��E)��Ӧ�¶ȸ���������ȥ������ʵ�������õ����ȥ�������ǿ�ȼ��Ϊ2.5����Ӧ�¶�1 060 �棬��Ӧʱ��60 min����Ӧȥ���ʿɴ�86.1%��
3.3 Ca(OH)2������Ӧ�����������
���ռ��ڿ�ȼ��Ϊ3.8ʱ����ͬ��Ӧʱ�����ռ���XRD��ͼ��ͼ4��ʾ���ֲ��Ŵ�ͼ��ͼ5��ʾ��
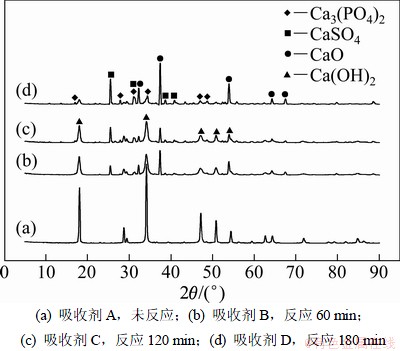
ͼ4 ��ȼ��Ϊ3.8ʱ��ͬ��Ӧʱ�����ռ���XRD��
Fig.4 XRD patterns of fresh sorbent and different reaction time sorbent at flow ratio of air to flue of 3.8
��2 ��ͬ�¶������ռ���P2O5��ȥ����
Table 2 Sorbent removal rate of P2O5 at different temperatures

ͼ5 ��ȼ��Ϊ3.8ʱ��ͬ��Ӧʱ�����ռ���XRD�ֲ��Ŵ�ͼ
Fig.5 Local microscope XRD patterns of different reaction time sorbent at flow ratio of air to flue of 3.8
��ͼ4��ͼ5���Կ�������Ӧ��ʼ�����ռ��������ַֽ�ΪCaO��ͬȼ�պ�β���е�������Ӧ����Ӧ����60 min���������(Ca3(PO4)2��PDF��09-0169)���淴Ӧ�Ľ���Ca3(PO4)2����ǿ��ӽ��������ӣ����뻯ѧ�������һ�¡���ͼ4��ͼ5���ɼ���Ca(OH)2�������ڷ�Ӧ����60 min��120 min��Ϊ���ԣ�180 min��ű������Ca(OH)2�����淴Ӧ�Ľ��У����ϵطֽ�ΪCaO���ͷų�H2O���γ��µĿ��ͱ��棻����ܵ��������ռ�����ȥ�����淴Ӧʱ��û�����Ե��½���
ͼ6��ͼ7��ʾ�ֱ�Ϊ��ͬ��ȼ�ȺͲ�ͬ��Ӧʱ�����������ռ���XRD���ֲ��Ŵ�ͼ���ɱ�2�ɼ���3�ַ�Ӧ������ռ������ӽ�����ȼ��Ϊ4.2��3.8ʱ��Ӧ����Ca3(PO4)2�����Ա�ͼ5(b)��ͼ7(a)���֣����ռ�C��Ca3(PO4)2������ǿ�ȱ����ռ�E�ĸߣ�˵�����²�������Ca3(PO4)2��������ɡ��ڿ�ȼ��Ϊ2.5��XRD���г�����Ca3(PO4)2�⣬�����������������ʽ�����(Ca5(PO4)3(OH)��PDF:84-1998)��ͬʱCaSO4�������IJ����ԡ�����Ander�ȵ��о�����[14]���������ܵ�ʱ�ᵼ��CaSO4�ķֽ⣬���ǰ��ķ������Զ϶���ȼ��Ϊ2.5ʱ������ȼ��������ȫ��Ӧ��
3.4 ���ռ�������ò�ֲ�����Ԫ�ط���
ͼ8��ʾ�ǵ���Ӧʱ��Ϊ60 min����ȼ�ȷֱ�Ϊ3.8��2.5ʱ��SEM��ɢ�������ͼ8��1��2��3��EDS��Ԫ�ط������3��ʾ��
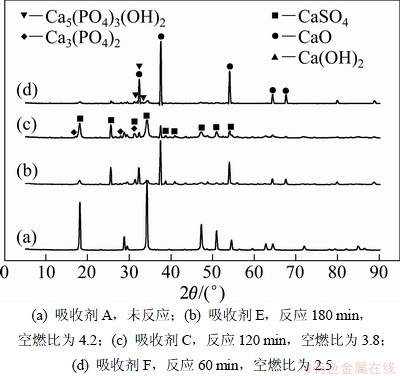
ͼ6 ��ͬ��ȼ�����������ռ���XRD��
Fig.6 XRD patterns of fresh sorbent and sorbent after reaction in different flow ratios of air to flue
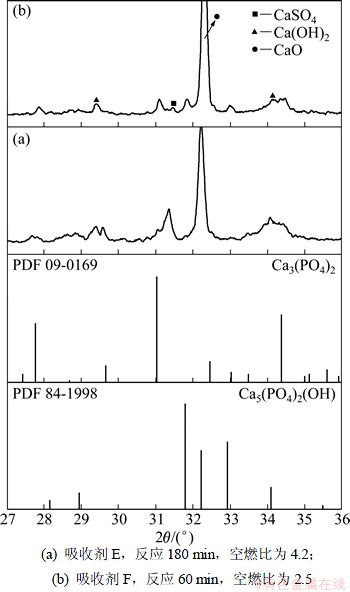
ͼ7 ��ͬ��ȼ�����������ռ���XRD�ֲ��Ŵ�ͼ
Fig.7 Local microscope XRD patterns of sorbent after reaction in different flow ratios of air to flue

ͼ8 ��ȼ��Ϊ3.8 (920 ��)��Ӧ60 min���ռ��Ϳ�ȼ��Ϊ2.5(1 060 ��)��Ӧ60 min���ռ���ɢ�������
Fig.8 Back-scattering electron images of reaction 60 min sorbent at flow ratio of air to flue of 3.8 (920 ��) and reaction 60 min sorbent at flow ratio of air to flue of 2.5 (1 060 ��)
��3 ���ռ�����EDSԪ�ط���(��������)
Table 3 EDS elemental analysis of sorbent surface %
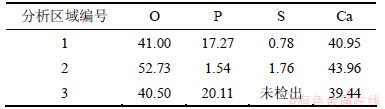
��ͼ8�ɼ����¶ȶ����ռ���ò����ҪӰ�죬 920 ��ʱ���������������ᾧ������(��ͼ8(a)��(b))��1 060 ��ʱ���������ʿ죬�ᾧ����С(��ͼ8(c)��(d))�������ͼ8(d)���Է��֣��ھ���������ϸ�ķ�϶�������ӱ�3���Կ�����������1��2��3�нᾧ��������ͷǽᾧ����Ca����������ͬ�����ᾧ��������1��3�������ֱ�ﵽ��17.27%��20.11%���ǽᾧ����2��ֻ��1.54%��˵���ᾧ��������������ף��¶����ӿ쾧���ٶȣ����¶�������������ȥ����
3.5 ����ѧ����
Ϊ��֤�������������ѧԤ�����֮���һ���ԣ�����ѧ���������������Ҫ�����������أ�
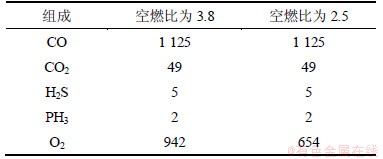
(1) ����ǰ���ʵ�鼰�������������β����ΪCO��CO2��PH3��H2S 4����֣�����β��ȼ����Ϊ1 m3(����Ӧ60 min)����ȼ��Ϊ2.5ʱ���ո�����ͬ����ȫ��Ӧ������������O2����������ȼ��Ϊ3.8ʱ�����ݿ�ȼ�ȼ���ó�O2�������������ʼ��������4��ʾ��
��4 ������������ɷּ���������
Table 4 Calculation of components and input values g
(2) ��������ѧ����ʱ�������ʾ��Ȼ�ϣ��������¶�920 �棬��1 060 ��ʱCaOͬP2O5��Ӧ�ļ���˹�����ܵ���CaOͬSO2��Ӧ�ļ���˹�����ܣ�ʵ������·�Ӧ��Ҫ���������ռ��������档Ca(OH)2�ļ��������ܰ�ʵ�ʼ��������㡣Ca(OH)2�ļ���������ǰ��ķ������������ʽ(2)~(5) ���м��㡣
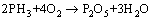 (2)
(2)
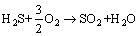 (3)
(3)
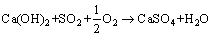 (4)
(4)
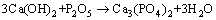 (5)
(5)
��ȼ��Ϊ3.8ʱ�������ͬ���ռ���Ӧ����CaSO4��Ca3(PO4)2��Ca(OH)2������Ϊ15.9 g����ȼ��Ϊ2.5ʱCaSO4�����ֽ⣬��ͬ���ռ���Ӧ������ֻ����Ca3(PO4)2��Ca(OH)2������Ϊ5.02 g��
(3) ��ѹ��101.3 kPa�����£����ݱ�2�ͱ�4���¶�920 ��ʱ�����������2 123 g(���㷶Χ0~2 123 g������43)��Ca(OH)2������15.9 g���¶�1 060 ��ʱ�����������1 835 g(���㷶Χ0~1 835 g������37)��Ca(OH)2������5.02 g��ͨ���ı�����������Է�Ӧ�������Ԥ�⣬�����ͼ9��ʾ��
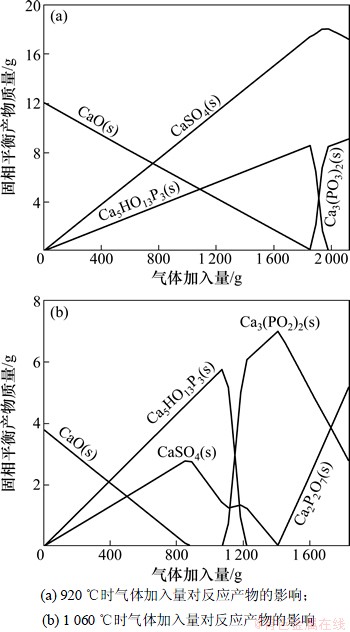
ͼ9 ��ͬȼ���¶�����ѧƽ�����ͼ
Fig.9 Thermodynamic equilibrium calculations in different combustion temperature
��ͼ9���Կ�������ͬ�¶��£����ռ����ȷֽ�ΪCaO��ͬP2O5��Ӧ����Ca5(PO4)3(OH)�����Ż����������������CaO�����Ĵ�������CaO��ȫ��Ӧ��֮��P2O5�����ͬCa5(PO4)3(OH)��Ӧ����Ca3(PO4)2������ʽ��ʾ��
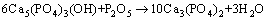 (6)
(6)
��ȼ���Ϳ�����ȫ��Ӧʱ��P2O5ͬ���ռ���Ӧ��������̬û���ܵ�Ӱ�죻��CaSO4ȴ��ò��ȶ������ڷֽ⡣ʵ������£����ռ���δ��ȫ��Ӧ�������ɲ�����ҪΪCa3(PO4)2�����ǰ��ķ�����֪����������Ca5(PO4)3(OH)�ľ������棬920 ��ʱ����������������������CaO�������㣬Ca5(PO4)3(OH)��Ѹ��ת��ΪCa3(PO4)2��1 060 ��ʱ���������ʿ죬���ռ��������ɴ����ľ���������������ռ�����ķ�Ӧ��ľ��ȣ��ӳ���Ca5(PO4)3(OH)����ʱ�䣻���������϶����2�����ص��£�һΪH2O�Ӿ������ͳ�����ΪCaSO4�ֽ⡣1 060 �������ѧ�����з�������Ca2P2O7������XRDͼ����δ�ҵ���Ӧ�������塣
Ϊ֤�����ڽ������(Ca2P2O7)������Co��(Co�в������������ͣ��������������Ч����)�Կ�ȼ��Ϊ2.5����Ӧ60 min������Ʒ����XRD���������Խ����ͼ10��ʾ��
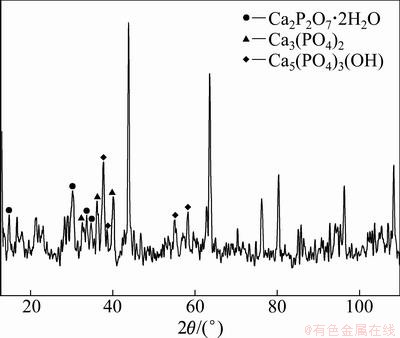
ͼ10 1 060 ��ʱ��Ӧ60 min������ƷXRD��
Fig.10 XRD pattern of particle sorbent after reaction at 1 060 �� for 60 min
��ͼ10���Է��֣���Ӧ�����ռ�����������Ca2P2O7��2H2O(PDF:41-0448)���ɣ������еĽᾧˮ��������Ʒ����ȴ��������¯�ڷ�Ӧ���ɵ�ˮ���ӽ�϶��ɡ��ɴ�֤��1 060 ��ʱCa3(PO4)2����ת��ΪCa2P2O7��ʵ����ͬ����ѧ������һ�¡�
4 ����
(1) ����������Ԥ��ȼ�ջ���β������ȼ��2.5ʱ�ﵽ����¶�1 060 �棬�ڿ�ȼ��1.7~4.2�ķ�Χ���¶����ȶ���870 �����ϣ��ﵽ������ȼú��������ȼ���¶ȡ�
(2) P2O5�������������ռ���870~1 060 �淶Χ�ھ��ܷ�Ӧ���ڷ�Ӧ������Ca(OH)2���Ϸֽ�����µĿ��ͱ��棬��180 min�ķ�Ӧʱ�������ռ���������ȥ����û�������½������ռ���������Ľᾧ����������P2O5���¶����������ڷ�Ӧ�Ľ��С�
(3) P2O5�������������ռ���Ӧ���γɼ�ʽ����ƣ�������P2O5�����ڼ�ʽ����ƾ�������ת��Ϊ����ơ����¶�Ϊ1 060 ��ʱ������Ƽ���ת��Ϊ������ơ�
�ο����ף�
[1] ���Ƽ�. ����β���ۺ�����[J]. ���븴�Ϸ�, 2008, 23(1): 45-48.
CHEN Shanji. Comprehensive utilization of tail gas from phosphorus furnace[J]. Phosphate and Compound Fertilizer, 2008, 23(1): 45-48.
[2] WANG Zhonghua, JIANG Ming, NING Ping, et al. Thermodynamic modeling and gaseous pollution prediction of yellow phosphorus production[J]. Ind Eng Chem Res, 2011, 50: 12194-12202.
[3] 2010��2011���й���������ҵ�о�����[R]. ����: ˮ��ľ���о�����, 2011: 7-8.
2010��2011 China phosphorite and phosphorous chemical industry report[R]. Beijing: PDAY Research, 2011: 7-8.
[4] ������. ���º�ѭ��������������չ����֮·. I: ����β�����ۺ��������״����սἼ��[J]. ���븴�Ϸ�, 2007, 22(3): 21-25.
YAN Minglang. Way of innovation and recycling economy for development of phosphorus chemical industry. I: Reclaiming,purification,and utilization of tail gas of yellow phosphorus[J]. Inorganic Chemicals Industry, 2007, 22(3): 21-25.
[5] ��. ���ڻ���β��������[J]. �����ι�ҵ, 2007(2): 9-12.
ZUO Jiangguo. On the utilization of yellow phosphorus tail gas[J]. Phosphate Industry, 2007(2): 9-12.
[6] ��Ԫ��, ��Դ, ������, ��. ����β��������¯�¼���[J]. ���ι�ҵ, 2010, 42(8): 39-40.
PEN Yuanhong, WAN Yuan, LIU Xinkun, et al. New steam boiler technology for utilization of yellow phosphorous tail gas[J]. Inorganic Chemicals Industry, 2010, 42(8): 39-40.
[7] ������, ����. ȼ����¯β����������¸�ʴ��ԭ��Ԥ����ʩ[J]. ұ����, 2007(4): 51-54.
HAN Mingjie, LIU Jinjing. Causes of low-temperature corrosion of gas-fired boiler end and preventive measure[J]. Metallurgical Power, 2007(4): 51-54.
[8] ������. ������չ���ߴ��º�ѭ������֮·. I: ����β�����վ�������[J]. ���ι�ҵ, 2009, 41(10): 2-6.
YAN Minglang. Way of innovation and recycling economy for development of phosphorus chemical industry. I: Reclaiming, purification, and utilization of tail gas of yellow phosphorus[J]. Inorganic Chemicals Industry, 2009, 41(10): 2-6.
[9] �κ���. ���й�����ҵ��չ��˼��[C]//�й���������(�˷�)��չ�߷���̳���й�������ҵ��ȹ����������ļ�. ����: ��ѧ��ҵ������, 2007: 19-21.
HE Haoming. China��s phosphate industry development[C]// China International Phosphorus Chemical (Xingfa) Development Summit Forum and China��s Phosphorus Chemical Industry Annual Conference. Beijing: Chemical Industry Press, 2007: 19-21.
[10] ��־��. ����¯β��Ϊȼ�ϵ�������¯��ʴԭ�����[J]. �����������幤��, 2011(3): 33-36.
SUN Zhili. Root cause analysis for corrosion of steam boiler fueled by off-gas from phosphorus furnace[J]. Sulphur Phosphorus and Bulk Materials Handling Related Engineering, 2011(3): 33-36.
[11] Anthony E J, Granatstein D L. Sulfation phenomena in fluidized bed combustion systems[J]. Progress in Energy and Combustion Science, 2001, 27: 215-236.
[12] Bale C W, Chartrand P, Degterov S A, et al. Factsage thermochemical software and database[J]. Calphad, 2002, 26(2): 189-228.
[13] Bale C W, Chartrand P, Degterov S A, et al. FactSage thermochemical software and databases-recent developments[J]. Calphad, 2009, 33(2): 295-311.
[14] Lyngfelt A, Langer V, Steenari B M, et al. Calcium sulphide formation in fluidized bed boilers[J]. The Canadian Journal of Chemical Engineering, 1995, 73(2): 228-233.
(�༭ ����ƽ)
�ո����ڣ�2012-02-11�������ڣ�2012-06-12
������Ŀ�����Ҹ����о���չ�ƻ�(��863���ƻ�)��Ŀ(2008AA062602)������ʡ��Ȼ��ѧ����������Ŀ(14051184)������������ѧ�������Ի���������Ŀ(2009-061��2009-050��2010136)
ͨ�����ߣ����ػ�(1957-)��Ů�����������ˣ���ʿ���´�����Ⱦ�о����绰��15877925068��E-mail��zhonghuakm@googlemail.com
ժҪ��ͨ��������Ԥ��ȼ�ջ���β������ȼ�չ����м����������ƣ��Ի���β��ȼ�ղ�����������������ȥ�����ı��ȼ�ȿ��췴Ӧ�¶ȶ�ȥ���ʵ�Ӱ�죬�����ռ�����XRD��SEM��EDS�����������Ԫ�ط���������FactSage6.2����ѧ���������ɵĹ���������Ԥ�⡣�о����������������Ԥ��ȼ�ջ���β��������¶�1 060 �棬�ڿ�ȼ��1.7~4.2�ķ�Χ��¯���¶����ȶ���870 �����ϡ����ռ���ͬ����������Ӧ�����������¶����߶����ӣ�1 060 ��ʱ������Ϊ86%��920 ��ʱ������Ϊ50%��870 ��ʱ������Ϊ30%����������ͬ���������������γɼ�ʽ����ƣ�֮���Ϊ����ƣ��ڸ����������ת��Ϊ������ơ���������Ԥ������ʵ����һ�¡�
[1] ���Ƽ�. ����β���ۺ�����[J]. ���븴�Ϸ�, 2008, 23(1): 45-48.
[3] 2010��2011���й���������ҵ�о�����[R]. ����: ˮ��ľ���о�����, 2011: 7-8.
[5] ��. ���ڻ���β��������[J]. �����ι�ҵ, 2007(2): 9-12.
[6] ��Ԫ��, ��Դ, ������, ��. ����β��������¯�¼���[J]. ���ι�ҵ, 2010, 42(8): 39-40.
[7] ������, ����. ȼ����¯β����������¸�ʴ��ԭ��Ԥ����ʩ[J]. ұ����, 2007(4): 51-54.
[8] ������. ������չ���ߴ��º�ѭ������֮·. I: ����β�����վ�������[J]. ���ι�ҵ, 2009, 41(10): 2-6.
[10] ��־��. ����¯β��Ϊȼ�ϵ�������¯��ʴԭ�����[J]. �����������幤��, 2011(3): 33-36.
