
J. Cent. South Univ. (2012) 19: 1219-1225
DOI: 10.1007/s11771-012-1132-0![]()
Crystal structures and theoretical investigation of anti-/syn-2,4-diphenylpentane- and 2,4-di-p-tolylpentane-2,4-diols
JIAO Yin-chun(������)1,2,3, CAO Chen-zhong(�ܳ���)2,3, ZHAO Xiao-lin(��С��)2,3
1. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China;
2. School of Chemistry and Chemical Engineering,
Hunan University of Science and Technology, Xiangtan 411201, China;
3. Key Laboratory of Theoretical Chemistry and Molecular Simulation of Ministry of Education,
Hunan University of Science and Technology, Xiangtan 411201, China;
? Central South University Press and Springer-Verlag Berlin Heidelberg 2012
Abstract:
2,4-diphenylpentane- and 2,4-di-p-tolylpentane-2,4-diols were investigated employing experimental and density functional theory (DFT) method at B3LYP/6-31G (d) level. The structure of syn-2,4-di-p-tolylpentane-2,4-diol (2b) was characterized by X-ray diffraction and compared with the crystal structures of anti- and syn-2,4-diphenylpentane-2,4-diols (1a and 1b). X-ray diffraction indicates that inter and intra-molecular hydrogen bonds are formed in the crystal structures. There is �ШC�� staking interaction in 1b and 2b. Good linear correlations and similar results are found between the experimental 1H and 13C NMR chemical shifts (��exp) and GIAO (Gauge Independent Atomic Orbital) method calculated magnetic isotropic shielding tensors (��calc). HOMO and LUMO molecular orbitals were calculated at the same levels with the different results. UV-vis absorption spectra of the compounds were recorded in EtOH, MeCN, n-BuOH and cyclohexane with different dielectric constants. It is found that the solvent effect is obvious when �� is 24.85(EtOH), 35.69(MeCN) and it is weak when �� is decreased to 17.33(n-BuOH), 1.18 (cyclohexane).
Key words:
1 Introduction
The 1,3-diol moiety is presented in a number of natural products with some biological activity and has attracted considerable attention in recent years. More specifically, 1,3-diols and its derivatives are important targets for many synthetic methodologies since they are high value chiral synthons [1-2] or building blocks [3] for the synthesis of many natural products, pharmaceuticals, agrochemicals, organic target compounds and chiral ligands in metal complexes [4-7]. The presence of hydroxyl groups gives opportunity for the interactions with a potential molecular target by donor or acceptor of hydrogen bonds. A series of linear aliphatic 1,3-diols and certain of their monoesters have been observed to be safe, readily metabolizable, and effective preservatives for raw grain and animal feeds. However, to the best of our knowledge, the biological activity of small molecule organic compounds containing the symmetric diaryl 1,3-diol units are very rarely reported. Many conventional methods for the preparation of 1,3-diols have been developed [8-10]. Recently, a series of symmetrical aromatic 1,3-diols were successfully synthesized from substituted aryl Grignard reagents and isopropenyl acetate in a one step reaction. The stereoselectivity and the mechanism of this reaction were performed using a combination of experimental and theoretical methods [11]. Since some of them are biologically active and widely used, an investigation of their structures and optical properties as probes for monitoring biochemical processes is useful.
2,4-diphenylpentane-2,4-diol and its derivatives (Fig. 1) show two important conformational isomers (anti- and syn-). X-ray structures of 1a and 1b were first reported [11]. Herein, X-ray structure of syn-2,4-di-p-tolylpentane-2,4-diol (2b) was reported and compared with 1a and 1b. The structure of 2a was optimized based on the crystal structure of 1a. For many diol derivatives, the detailed absorption spectra characteristics and quantum chemical calculations are not available [12]. The purpose of this work is to investigate the crystal structures of 1a, 1b and 2b and perform an initial study of the full electronic absorption spectra in different solvents for the titled compounds and TDDFT calculations. This combined study will facilitate the understanding of the detailed structural and property features of serial of aryl 1,3-diols and contribute to the further information on the molecular level.

Fig. 1 Structures of two anti- (a) and syn- (b) conforms
2 Experimental
2.1 Synthesis of materials
The synthesis of all chemicals and NMR spectra can be found in Ref. [11].
2.2 X-ray crystallography
Crystals suitable for X-ray diffraction were grown from petroleum-ethyl acetate (3:1). The crystallographic analysis was performed on a Smart CCD Apex II diffractometer with Mo K�� monochromated radiation (��=0.710 73 ?) at 296 K. Empirical absorption correction was applied. The structure was solved by the direct method and refined by the full-matrix least-squares method on F2 using the SHELXL-97 software [13]. All of the non-hydrogen atoms were refined anisotropically. The hydrogen atoms were generated geometrically. CCDC 822128 (Fig. 2) contains the supplementary crystallographic data for this work. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/ conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44-1223-336033).
2.3 UV-vis absorption procedure
Electronic absorption spectra in solution were recorded on a LAMBDA-35 UV-vis spectrometer in the concentration range from 10-3 to 10-5 mol/L. The solvents used in absorption experiments (ethanol, acetonitrile, 1-butanol and cyclohexane) were of spectroscopic grade and used as purchased.

Fig. 2 Structure of 2b from X-ray diffraction
2.4 Computational procedure
The starting atomic coordinates were taken from the final X-ray refinement cycle. The geometry optimizations were performed at the B3LYP/6-31G (d) level with the Gaussian 03 program package [14]. The 1H and 13C chemical shifts together with the shift of tetramethylsilane (TMS) were fully optimized and were calculated on the same basis of GIAO method [15-17]. The calculated absolute shielding of the molecule was defined as the absolute shielding returned by the program in chemical shifts subtracting the absolute shielding of TMS (��calc=��abs-��absTMS). Vibration frequencies calculated ascertain the structures to be stable (no imaginary frequencies) for the investigated forms. Electronic spectrum was calculated by TDDFT method. The PCM solvent model was used in the Gaussian calculations with ethanol, acetonitrile, n-butanol and cyclohexane as the solvents.
3 Results and discussion
3.1 Hydrogen bond description
In the crystal lattice of 1a, 1b [11] and 2b, there are two potentially intramolecular and intermolecular hydrogen bonds (HBs) [18-19] interactions (O��H������O). The hydrogen bond parameters of 2b are listed in Table 1.��ʾ��Ӧ�������ַ���ƴ��
Table 1 Hydrogen bond parameters in crystal structure of 2b

The distances and angles between donor and acceptor of 2b are 2.67(2) ?, 158�� for O1��H(1)������O(2), and 2.80(2) ?, 172�� for O(2)��H(2)������O(1) (symmetry code: -x, -y, -z), respectively. In 1a, 1b and 2b, intramolecular and intermolecular O�CH������O hydrogen bonds form centrosymmetric dimers (Fig. 3) [20]. Both types of hydrogen bonds are asymmetric and angular. However, the distance between the proton donor (O��H) and proton acceptor (H������O) atom falls in the range of 2.50-2.90 ?, and characteristic of stronger hydrogen bonds can be found in many biological systems [21].

Fig. 3 Crystal packing and O��H������O hydrogen bonds of 2b obtained by X-ray diffraction
The most fascinating structural feature is that the dihedral angle between the two benzene rings of anti-1a is 134.92�� and the angles of syn-1b, 2b are 20.04�� and 13.72��, respectively. Furthermore, in 1b and 2b, intramolecular face-to-face �ШC�� staking interactions [22] are found with the length of 3.69 and 3.54 ? for the two benzene rings. There is also edge-to-face (C��H����) arrangements with the length of 3.30 and 3.18 ? between any hydrogen atom and the centroid of the C6 ring of the adjacent molecular in 1b and 2b [23].
3.2 Structure analysis
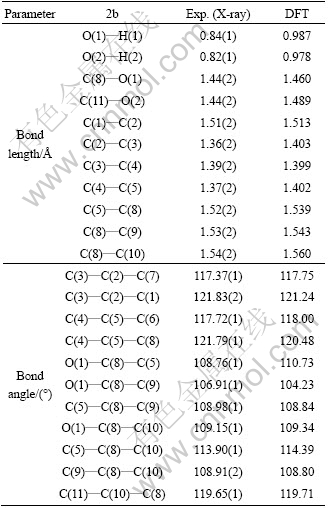
Summaries of selected bond lengths and angles of 2b are given in Table 2. The largest deviation of the DFT optimized bond-lengths/angles is O��H bond about 0.82-0.84 ? (X�Cray) [24] and 0.98-0.99 ? (DFT) [25]. Calculated bond distance in the isolated molecule is slightly larger than that determined by X-ray diffraction [26]. The small differences can be attributed to the fact that the theoretical calculations were carried out with isolated molecules in the gaseous phase whereas the experimental values were based on molecules in the crystalline state. On the other hand, only the monomeric unit was optimized, while in the solid state the compounds occur in the form of the hydrogen-bonded dimers.
Table 2 Selected bond length, and bond angle of 2b
The results of NMR of 2b compared with the GIAO isotropic magnetic shielding tensors (��calc) are depicted in Table 3. Resonances of 2b show the important difference: the two hydrogen of CH2 (C-10) of 2b (syn-isomer) split into four peaks as 2.55��10-6, 2.52��10-6, 2.41��10-6, 2.38��10-6 in 1H NMR while no split peaks in 2a (anti-isomer). The homogeneous results are presented in 1a and 1b. The chemical shift of the two OH groups in 2b is 3.59��10-6 (for 1a, 2a and 1b, 3.89��10-6, 3.63��10-6 and 3.78��10-6, respectively), however in calculations they are 0.84��10-6 and 5.42��10-6 respectively. Difference of chemical shift may be attributed to the formation of intramolecular and intermolecular hydrogen bonds. Plotting the experimental 1H and 13C chemical shifts (��exp) vs the ��calc for all species except for O������H, a linear regression is obtained: ��exp=a����calc+b (Table 3). As observed, the correlation between experimental chemical shifts and calculated isotropic screening constants for 13C (R2= 0.996) is better than that for 1H (R2=0.964). The results obtained from the optimization show reasonable agreement with experimental results as seen.
Table 3 Experimental 1H and 13C chemical shift (��exp/10-6) and calculated GIAO magnetic isotropic shielding tensor (��calc/10-6) (��exp=a����calc+b)) for 2b

3.3 Frontier molecular orbitals analysis
HOMO and LUMO molecular orbitals were calculated for 2a and 2b with the B3LYP/6-31G (d) basis (Fig. 4). In these structures, HOMO and LUMO of 2a are delocalized on half molecule, giving rise to two separated fragments, because of the electronic density distributed in the part of the benzene. HOMO and LOMO of 2b are delocalized on the hole molecule, and the LUMO orbital has a clear ��* characteristic, localized at the benzene ring. The frontier molecular orbitals of 1a and 1b are similar with 2a and 2b. The HOMO-LUMO energy gap of 2b in vacuum is 0.215 7 eV (Table 4). The greater the energy difference between HOMO and LUMO orbital, the more difficult the electronic transition, and the more the stable compounds.

Fig. 4 Computed frontier orbitals of 2a and 2b
Table 4 Calculated EHOMO, ELUMO, ��E(ELUMO-EHOMO) and dipole moment (��/D)

3.4 UV-vis spectra
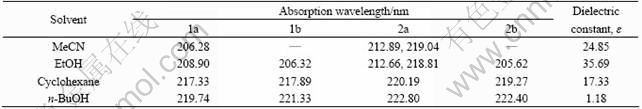
Experimental UV-vis spectra and main absorption wavelength of title compounds are shown in Fig. 5 and Table 5. The solvents, including classification and solvent descriptors, protic solvents (EtOH, n-BuOH), electron pair donating solvents (CH3CN), and that with no specific solvent-solute interactions (cyclohexane) were used [27]. For 2a, the absorption spectrum has two more intense peaks (212.89 nm, 219.04 nm and 212.66 nm, 218.81 nm) in EtOH and MeCN. No apparent absorption peaks are found for 1b and 2b in MeCN. Both of 1b and 2b absorption bands have an apparent blue shift in CH3CN and have a slight blue shift in EtOH. In n-BuOH and cyclohexane, no significant trend is observed. In comparison with the maximum absorption bands of the titled compounds, those of the syn-isomers are red shifted in n-butanol and cyclohexane, which is mainly attributed to a slight lower energy gap of 0.228 0 eV (1a), 0.219 9 eV (1b), 0.222 0 eV (2a) and 0.215 7 eV (2b), as listed in Table 5. In high dielectric constant solvents (MeCN and EtOH), the bands are broad and smooth with smaller absorption; in lower dielectric constants solvents (n-BuOH and cyclohexane), the absorption has sharp peak shape. It is found that the solvent effect is obvious when �� is 24.85 (EtOH), 35.69 (MeCN) and is weak when �� is decreased to 17.33 (n-BuOH) and 1.18 (cyclohexane).

Fig. 5 Experimental absorption spectra of 1a, 1b, 2a and 2b in four solvents: (a) In MeCN; (b) In EtOH; (c) In n-BuOH; (d) In cyclohexane
Table 5 Experimental absorption spectra wavelength of 1a, 1b, 2a and 2b in four solvents
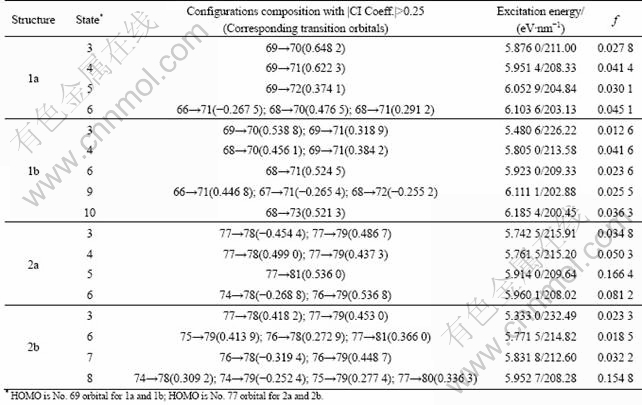
In order to understand electronic transitions of compounds, TDDFT calculations on electronic absorption spectra in different solvent environments were performed by using PCM model on the B3LYP/6-31G(d) level. The similar results are found for the same isomers. The calculated maximum absorption wavelengths and oscillator strengths for 2a and 2b in CH3CN are given in Table 6. The calculation is modeled based upon the solvent dielectric results in minimal solvatochromic shifts. On the basis of the calculated results, the electronic transition of anti-isomers is mainly from HOMO to other LUMOs and that of syn-isomers is mainly from different HOMOs to other LUMOs. The absorption band is derived from HOMO��LUMO, HOMO��LUMO+1, HOMO��LUMO+2 for 1a and HOMO��LUMO, HOMO��LUMO+1, HOMO�� LUMO+3 for 2a. The syn-isomers for 1b is HOMO �� LUMO, HOMO��LUMO+1, HOMO�C1��LUMO, HOMO�C1��LUMO+1 and HOMO��LUMO, HOMO�� LUMO+1, HOMO�C2��LUMO+1, HOMO��LUMO+3, HOMO�C1��LUMO+1, HOMO�C1��LUMO+2 for 2b. Based on the compositions of the molecular orbitals, it can be concluded that the main absorbance band is originated from ��aryl����aryl* and �ҡ���aryl* electronic transitions of the titled compounds.
Compared with the results of experiment, the results of TDDFT in four solvents have the similar results. The discrepancy between experiment and TDDFT calculations may be attributed to two aspects [28] which involve DFT and solvent effect. So, it is very difficult to make the theoretical calculation consistent with the experimental data quantitatively. Though the discrepancy exists, the TDDFT calculations are capable of describing the spectral features of the titled compounds as compared with experiment.
Table 6 Computed excitation energies, electronic transition configurations and oscillator strengths (f) for optical transitions with f>0.01 of absorption bands in UV-vis region for 1a, 1b, 2a and 2b in MeCN
4 Conclusions
1) The geometries of the 1a, 1b and 2b molecules are influenced by crystal packing forces such as intermolecular and intramolecular hydrogen bonding contacts. There are �ШC�� stacking interactions of the aromatic rings in syn-isomers.
2) The calculated magnetic isotropic shielding tensors (��calc) have good linear correlations with experimental 1H and 13C chemical shifts (��exp).
3) The UV�Cvis absorbance of the titled compounds exhibits solvent-dependent effect. The solvent effect is obvious when �� is 24.85 (C2H5OH), 35.69 (CH3CN) and is weak when �� is decreased to 17.33 (n-BuOH), 1.18 (cyclohexane). TDDFT calculations are mainly ascribed to �ШC��* and ��-��* transitions.
References
[1] ICHIBAKASE T, NAKAJIMA M. Direct enantioselective aldol-Tishchenko reaction catalyzed by chiral lithium diphenylbinaphtholate [J]. Organic Letters, 2011, 13(7): 1579-1581.
[2] ZACHARIA J T, TANAKA T, HAYASHI M. Facile and highly enantioselective synthesis of (+)- and (-)-fluvastatin and their analogues [J]. The Journal of Organic Chemistry, 2010, 75(22): 7514�C7518.
[3] TROET B M, SHIN S, SCLAFANI J A. Direct asymmetric Zn-Aldol reaction of methyl vinyl ketone and its synthetic applications [J]. Journal of the American Chemical Society, 2005, 127(24): 8602-8603.
[4] RYCHNOVSKY S D. Oxo polyene macrolide antibiotics [J]. Chemical Reviews, 1995, 95(6): 2021-2040.
[5] MARINO J P, MCCLURE M S, HOLUB D P, COMASSETO J V, TUCCI F C. Stereocontrolled synthesis of (-)-macrolactin A [J]. Journal of the American Chemical Society, 2002, 124(8): 1664-1668.
[6] YUI N. Supramolecular design for biological applications [M]. Boca Raton, New York: CRC Press, 2002: 1-401.
[7] MATSUO J I, HATTORI Y, ISHIBASHI H. Br?nsted acid catalyzed asymmetric reduction of ketones and acyl silanes using chiral anti-pentane-2,4-diol [J]. Organic Letters, 2010, 12(10): 2294-2297.
[8] RAMACHANDRAN P V, CHATTERJEE A. Gem- difluorinated homoallyl alcohols, ��-hydroxy ketones, and syn- and anti-1,3-diols via ��,��-difluoroallylboronates [J]. Organic Letters, 2008, 10(6): 1195-1198.
[9] CHEN K, RICHTER J M, BARAN P S. 1,3-Diol synthesis via controlled, radical-mediated C-H functionalization [J]. Journal of the American Chemical Society, 2008, 130(23): 7247-7249.
[10] CHO H Y, MORKEN J P. Ni-catalyzed corylative diene-aldehyde coupling: The remarkable effect of P(SiMe3)3 [J]. Journal of the American Chemical Society, 2010, 132(22): 7576-7577.
[11] JIAO Yin-chun, CAO Chen-zhong, ZHOU Zai-chun. Direct synthesis of anti-1,3-diols through nonclassical reaction of aryl grignard reagents with isopropenyl acetate [J]. Organic Letters, 2011, 13(2): 180-183.
[12] NILEWSKI C, GEISSER R W, EBERT M O, CARREIRA E M. Conformational and configurational analysis in the study and synthesis of chlorinated natural products [J]. Journal of the American Chemical Society, 2009, 131(43): 15866-15876.
[13] SHELDRICK G M. SHELXTL-97 [D]. G?ttingen, Germany: University of G?ttingen, 1997.
[14] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 03 [M]. Pittsburgh, PA: Gaussian Inc, 2003.
[15] WOLINSKY K, HINTON J F, PULAY P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations [J]. Journal of the American Chemical Society, 1990, 112(23): 8251-8260.
[16] BENASSI R, FERRARI E, LAZZARI S, SPAGNOLO F, SALADINI M. Theoretical study on curcumin: A comparison of calculated spectroscopic properties with NMR, UV�Cvis and IR experimental data [J]. Journal of Molecular Structure, 2008, 892(1-3): 168-176.
[17] DEMIR S, DIN?ER M, KORKUSUZ E, YlLDlRlM ?. An experimental and theoretical approach to molecular structure of 4-benzoyl-5-phenyl-1-p-methoxyphenyl-1H-pyrazole-3-carboxylic acid methanol solvate [J]. Journal of Molecular Structure. 2010, 980(1): 1-6.
[18] SOBCZYK L, GRABOWSKI S J, KRYGOWSKI T M. Interrelation between H-bond and Pi-electron delocalization [J]. Chemical Reviews, 2005, 105(10): 3513-3560.
[19] YANG Xin-guo, YUAN Huan, ZHAO Qiu-li, YANG Qing, CHEN Xian-hong. Self-assembly constructed by perylene bisimide derivatives bearing complementary hydrogen-bonding moieties [J]. Journal of Central South University of Technology, 2009, 16: 206-211.
[20] HOJNIAK A S, DEPERASI?SKA I, JERZYKIEWICZ L, SOBOTA P, HOJNIAK M, PUSZKO A, HARASZKIEWICZ N, ZWAN G, JACQUES P. Crystal structure, spectroscopic, and theoretical investigations of excited-state proton transfer in the doubly hydrogen-bonded dimer of 2-butylamino-6-methyl-4-nitropyridine N-Oxide [J]. The Journal of Physical Chemistry A, 2006, 110(37): 10690-10698.
[21] DZWOLAK W, RAVINDRA R, WINTER R. Hydration and structure�Cthe two sides of the insulin aggregation process [J]. Physical Chemistry Chemical Physics, 2004,6(8): 1938-1943.
[22] G??WKA M L, MARTYNOWSKI D, KOZ?OWSKA K. Stacking of six-membered aromatic rings in crystals [J]. Journal of Molecular Structure, 1999, 474(1): 81-89.
[23] RUSSELL V, SCUDDER M, DANCE I. The crystal supramolecularity of metal phenanthroline complexes [J]. Journal of the Chemical Society, Dalton Transactions, 2001: 789-799.
[24] CHANDRASEKHAR V, NAGENDRAN S, BOOMISHANKAR R, BUTCHER R. The effect of sterically hindered ligands on the solid-state structures of organosilanediols containing Si-N bonds [J]. Inorganic Chemistry, 2001, 40: 940-945.
[25] LUQUE F J, L?PEZ J M. Role of intramolecular hydrogen bonds in the intermolecular hydrogen bonding of carbohydrates [J]. The Journal of Physical Chemistry A, 1998, 102(33): 6690-6696.
[26] WANG Xian-long, MALLORY F B, MALLORY C W, Beckmann P A, Rheingold A L, Francl M M. CF3 rotation in 3-(trifluoromethyl) phenanthrene. X-ray diffraction and ab initio electronic structure calculations [J]. The Journal of Physical Chemistry A, 2006, 110(11): 3954-3960.
[27] BAUGHMAN B M, STENNETT E, LIPNER R E, RUDAWSKY A C, SCHMIDTKE S J. Structural and spectroscopic studies of the photophysical properties of benzophenone derivatives [J]. The Journal of Physical Chemistry A, 2009, 113(28): 8011-8019.
[28] ZHANG Cai-rong, WU You-zhi, CHEN Yu-hong, CHEN Hong-shan. Geometries, electronic structures and related properties of organic dye sensitizers JK16 and JK17 [J]. Acta Physico-Chimica Sinica, 2009, 25(1): 53-60.
(Edited by YANG Bing)
Foundation item: Projects(21072053, 20772028) supported by the National Natural Science Foundation of China; Projects(10K025, 11C0527) supported by the Scientific Research Fund of Hunan Provincial Education Department, China; Project(LKF0901) supported by the Open Foundation of Key Laboratory of Theoretical Chemistry and Molecular Simulation of Ministry of Education, Hunan University of Science and Technology, China
Received date: 2011-10-20; Accepted date: 2011-12-20
Corresponding author: CAO Chen-zhong, Professor, PhD; Tel: +86-731-58291336; E-mail: czcao@hnust.edu.cn
Abstract: 2,4-diphenylpentane- and 2,4-di-p-tolylpentane-2,4-diols were investigated employing experimental and density functional theory (DFT) method at B3LYP/6-31G (d) level. The structure of syn-2,4-di-p-tolylpentane-2,4-diol (2b) was characterized by X-ray diffraction and compared with the crystal structures of anti- and syn-2,4-diphenylpentane-2,4-diols (1a and 1b). X-ray diffraction indicates that inter and intra-molecular hydrogen bonds are formed in the crystal structures. There is �ШC�� staking interaction in 1b and 2b. Good linear correlations and similar results are found between the experimental 1H and 13C NMR chemical shifts (��exp) and GIAO (Gauge Independent Atomic Orbital) method calculated magnetic isotropic shielding tensors (��calc). HOMO and LUMO molecular orbitals were calculated at the same levels with the different results. UV-vis absorption spectra of the compounds were recorded in EtOH, MeCN, n-BuOH and cyclohexane with different dielectric constants. It is found that the solvent effect is obvious when �� is 24.85(EtOH), 35.69(MeCN) and it is weak when �� is decreased to 17.33(n-BuOH), 1.18 (cyclohexane).
