
���±�ţ�1004-0609(2009)10-1835-05
�����仯����Mg2Pb�ĵ��ӽṹ�͵�������
���������� �£���������³ ����������
(����������ѧ ����ʡ�²����Ʊ���ӹ��ص�ʵ���ң����� 650093)
ժ Ҫ�����õ�һ��ԭ�����������˽����仯����Mg2Pb�ĵ��ӽṹ�Լ��������ʣ�����Voigt-Reuss-Hill��������õ�Mg2Pb�ĵ���ģ�����б�ģ�������������Mg��Pb��̬�ܶȵĹ�����Ҫ��Mg��2p�����Pb��5d��������ΪMg��3s�����Pb��6p�����Pb��6s���������С����Mgԭ����Χ�д����ĵ�ɴ��ڣ��ʵ��͵Ľ�����������Mg��Pb֮����ڹ��õĵ�ɣ��н�ǿ�������ԣ��Թ��ۼ���ʽ���ڣ��������ɵĻ��䲻�ʹ��ۼ���ռ�������٣���������ռ�����ϴ�Mg2Pb������ʰ�����ԣ�Mg2Pb�ĵ���ģ�����б�ģ���ֱ�Ϊ68.6��27.9 GPa��Pugh�����оݺͲ��ɱȾ�����Mg2Pb���д��ԡ�
�ؼ��ʣ�
��һ��ԭ����Mg2Pb�����ӽṹ������������
��ͼ����ţ�TB331 ���ױ�ʶ�룺A
Electronic structure and elastic properties of intermetallics Mg2Pb
DUAN Yong-hua, SUN Yong, PENG Ming-jun, LU Li, ZHAO Ru-long
(Key Lab of Advanced Materials of Yunnan Province, Kunming University of Science and Technology,
Kunming 650093, China)
Abstract: The electronic structure and the elastic properties of Mg2Pb were investigated by the first-principles method. The elastic modulus and shear modulus for Mg2Pb were calculated from the theoretical elastic constants by Voigt-Reuss-Hill averaging scheme. The results show that the major contribution to DOS of Mg and Pb are the 2p orbit of Mg and the 5d orbit of Pb, followed by the 3s orbit of Mg and the 6p orbit of Pb, the 6s orbit of Pb is the smallest one. There are a large number of charges around Mg, it has the characteristics of typical metal bond. Mg and Pb share some charges to form covalent bond, but the distortion of the charge at the junction is little; the proportion of covalent bond is less than the metal bond, Mg2Pb is semimetal. The elastic modulus and shear modulus of Mg2Pb are 68.6 and 27.9 GPa, respectively. Based on Pugh empirical criterions and Poisson��s ratio, Mg2Pb is brittle in nature.
Key words: first-principle; Mg2Pb; electronic structure; elastic properties
Ǧ�ڳ����¾ͻ�����ظ����ٽᾧ����ͳ��ǿ������[1]�����ǿ�����ӹ�Ӧ��ǿ����Ǧ����Ͻ����á���ˣ�Ϊ�˻��ǿ�Ⱥ�Ӳ�Ƚϸߵ�����Ǧ���Ͻ�����ṹ����һ�廯��Ҫ�����Ʊ���һ�����͵�Mg/Pb�Ͻ������Խ����仯����Ϊ���IJ��ϡ����ڽ����仯�����в�ͬ����ԭ�ӵ�ԭ�Ӽ�ǿ�������Ե���λ�������˶���ʹ�ò��ϵ�ǿ�ȴ������[2]���йؽ����仯��������������о�����[3]���ڶ��������仯�����У���ɼ��ǽ������빲�ۼ��Ļ�ϡ�FOX��TABBERNOR[4]���о�֤�����ڦ�-NiAl�еļ��Ϸ�ʽ�ǹ��ۼ��ͽ��������档YOO��FU[5]���û��ھ����ܶȷ������۵ĵ�һ��ԭ��������Ni3Al�����仯����ĵ��ӽṹ���㣬���ý��Ҳ֤�����ڽ����仯��������ž������������ԵĹ��ۼ����á�
������Խ����仯����Mg2Pb�ĵ��ӽṹ�о����١�van DYKE��HERMAN[6]��������ƽ�沨���Ʒ���Mg2Pb������ܴ��ṹ������̽�֣���δ���о���ɼ�����van ATTEKUM��[7]�о���һЩ����CaF2�ṹ�Ľ����仯����ļ۴��ṹ�����ת�Ƶȣ���Ҫ��չ��AuX2(X=Al, Ga, In)��Mg2X(X=Si, Ge, Sn)����ع�����PETER[8]�Ի�����Mg2Si��Mg2Ge�ĵ��ӽṹ�������о��������漰�Ͻ�Mg2(Sn)x(Pb)1-x�İ�������ĵ������Ѩ�����棻WOOD��ALEX[9]�о��˷�өʯ�ṹ�İ뵼��Mg2Si�ĵ���ܶ����ܴ��ṹ��
��ˣ�Ϊ���о������仯����Mg2Pb��Mg/Pb�Ͻ���ѧ����Ӱ������ۣ����õ�һ��ԭ�����㷽��ϵͳ���о���Mg2Pb���ܴ��ṹ������ܶȡ�Mg2Pb�ļ��������Լ��������ʡ�
1 ���㷽����ģ��
���û����ܶȷ������۵�ƽ�沨���Ʒ���[10]���ӵ�һ��ԭ��������ѡ������ݶȽ���(GGA)�µ�PBE(Perdew Burke-Ernzerhof)����[11]���������������ܣ�ѡ���������ڵ����ӿռ��н��м���[12]���ڼ����У�ƽ�沨��ֹ��ȡ440 eV��Brillouin����K��ȡΪ4��4��4���ڴ������ӳ�ԥʱ���ù����ݶȷ������ܶȻ�Ϸ���[13]���ṹ�Ż�����ʱ����������������1��10-5 eV/atom��ÿ��ԭ���ϵ�������0.3 eV/nm������ƫ��Ϊ1��10-4 nm��Ӧ��ƫ��Ϊ0.05 GPa����Ǣ����Ϊ1��10-6 eV/atom�����м��㹤������CASTEP (Cambridge serial total enery package)ģ��[14]����ɡ�
Mg2Pb���з�өʯ�ṹ(Anti-CaF2��cF12)���ռ�ȺΪFm3m����������a=6.836 ?��Pbԭ��ռ�ݵ�(0��0��0)��Mgԭ�ӷֱ�ռ�ݵ�a/4(1,1,1)�͵�3a/4(1, 1, 1)[15]���侧��ģ����ͼ1��ʾ��
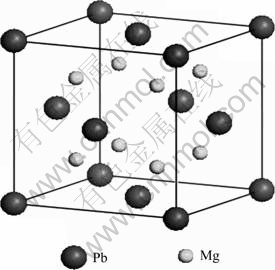
ͼ1 Mg2Pb�ľ���ģ��
Fig.1 Cell model of Mg2Pb
2 ������������
Ϊ���о�Mg2Pb����ṹ��ԭ�Ӽ�Ķ̳�����ã�������Mg2Pb���ܴ��ṹ������̬�ܶȡ�����̬�ܶȡ����Ƽ������漰��Mg:2p63s2��Pb:5d106s26p2��������̬�ܶ��еķ�̬�ܶȺ���������f����Ĺ��ס��ܴ��ṹ��̬�ܶ���ͼ2��ʾ����ͼ2�п�֪��Mg2Pb����һ���ĵ����ԣ�������[16]��Ϊ���ϣ�ͼ�������ԵĴ�϶������[6]����Ĵ�϶ֵ0.15 eV��С�����Ӻ��������������Ծ�����������磬��˶��߽������һ�¡��ܴ��ṹͼ����Mg2Pb�ļ۴������Ͽ��Է�Ϊ4������-10~-8 eV���¼۴�����-5~0 eV���ϼ۴������Լ�λ��-16 eV����Ϊ3 eV�ļ۴�����-45~-43 eV�ļ۴���������λ��0~18 eV(��ͼ2(a))��̬�ܶȷֲ�ͼ�п��Կ�����λ��-16 eV����Ϊ3 eV�ļ۴�����d������ף���-45~-43 eV�ļ۴�������p�������(��ͼ2(b))��
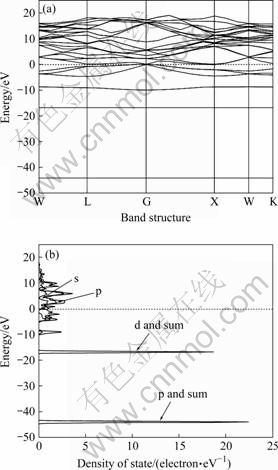
ͼ2 Mg2Pb�ܴ��ṹ��̬�ܶȷֲ�
Fig.2 Band structure and DOS of Mg2Pb: (a) Band structure; (b) Density of states
Ϊ�˷���Mg��Pb��̬�ܶȵIJ�ͬ���ף�������Mg2Pb��Ӧԭ�ӵķֲ�̬�ܶȺ���̬�ܶȣ���������ͼ3����Ȼ��Mg2Pb���ϼ۴�����Mg��2p�����Pb��6p����γɵģ�Mg��3s����в��ֹ��ף���Pb��5d�����6s�����û�й��ס��¼۴�����Mg��3s�����Pb��6s�����ͬ�����ɣ�Mg��2p���Ҳ�в��ֹ��ס�-45~-43 eV�ļ۴�����Mg��2p������ģ�λ��-16 eV���ļ۴�����Pb��5d������ģ��������۴������к�ǿ�ľ����ԣ����ڴ������۴����������ϡ��������۴�֮�������ý�������Mg2Pb����������Ӱ�첻�������֣���Ҫ��Դ��Mg��3s��2p������ס���ͼ3��֪�����ϼ۴���(-5~0 eV)��Mg��2p�����Pb��6p����ص��ϴ��������ڴ���������Ϊǿ�ҵĹ���ӻ���ͬ���ģ�Mg��3s��Pb��6s���¼۴���(-10~-8 eV)��һ������ӻ���Mg��3s��2p����ڵ�����Ҳ���ڹ���ӻ���

ͼ3 Mg2Pb�ķ�̬�ܶ�����̬�ܶ�
Fig.3 Partial and total DOS of Mg2Pb
Ϊ���˽���ϵ�ĵ�ɷֲ��͵��ת�����������Mgԭ����Pbԭ�ӵijɼ���ʽ����Mg2Pb(110)��ĵ���ܶ��Լ���ֵ���ܶȽ��м��㡣��ֵ���ܶ�Ϊ
![]()
ʽ�У���sΪ������ϵ�ĵ���ܶȣ���Mg�ͦ�Pb�ֱ�ΪMgԭ����Pbԭ�ӵĵ���ܶȡ�
ͼ4��ʾΪ�����仯����Mg2Pb(110)��ĵ���ܶȺͲ�ֵ���ܶ�ͼ������[7]������Mg2Pb�У�Pbԭ�Ӵ�Mg�õ�0.40�����ӣ����Pb��6p������ʸ����ԣ�Mg������ɡ��ڵ���ܶȷֲ�ͼ�У��γ����Ӽ�ʱ��������Ӽ��У����γɹ��ۼ�ʱ�������ɼ�������[17]����ͼ4(a)�ɼ���ÿһ��ԭ����Χ�ĵ�ɳ����ηֲ�����Mgԭ����Χ�д����ĵ�ɴ��ڣ��ʵ��͵Ľ�����������ͼ4(b)��������Pbԭ��λ�ã�����ܶȲ�Ϊ��ֵ������Mg��Pb֮��ĵ���ܶȲ�Ϊ�ϴ����ֵ��˵��Mg��Pb���ڶ��߹��õĵ�ɣ�Mg-Pbԭ�Ӽ��γ��˷����Խ�ǿ�Ĺ��ۼ�����̬�ܶȷ�����֪��Mg��Pb���ڹ���ӻ�����Mg��Pb֮��ĵ�����ֻ�в����ص��������ɵĻ��䲻�ʹ��ۼ���ռ�������٣���������ռ�����ϴ�Mg2Pb������ʰ�����ԣ�������������[18]��

ͼ4 Mg2Pb(110)�����ܶ�ͼ���ֵ���ܶ�ͼ
Fig.4 Charge densities (a) and charge densities difference (b) on (110) plane of Mg2Pb
��������ӽṹͬ���ļ��㷽�����м��㡣ƽ�沨�ض���ȡ440 eV�����ռ���k���ľ���ѡΪ0.4 nm-1����Ǣѭ���������������ֵΪ1��10-6 eV/atom����ԭ�Ӽ������������0.01 eV/nm�����ȶ�Mg2Pb�������о�������ԭ��λ���Ż�������õ���ƽ�⾧����Ϊ6.908 ?����ʵ��ֵ6.836 ?����С�������Ż����Mg2Pb���������䵯�Գ�����������1���У���ֵ��ʵ��ֵ�����Ǻϡ����Գ�����������������仯����Ľṹ�ȶ��ԣ������������壬�䵯�Գ���Ӧ����ѭ��������[21]��C44��0��C11��|C12|��C11+2C12��0���ɱ�1����õ��ĵ��Գ�����������������������Mg2Pb�ľ���ṹ���ȶ��ġ�
��1 Mg2Pb����ĵ��Գ���
Table 1 Calculated elastic constants for Mg2Pb(GPa)

ͨ�����Գ������Լ���Mg2Pb��ƽ����ģ��K[22]������ģ��E[23]���б�ģ��G[24]�Ͳ��ɱȦ�[23]�ȵ������ʡ�����������ϵ�����㹫ʽ�ֱ�Ϊ

ʽ(6)��(7)�У�SijΪCij���������ģ��E�Ͳ��ɱȦ͵ļ��㹫ʽ���£�
![]()
![]()
����������ʽ�������Mg2Pb����ĵ���ģ�����б�ģ������ģ���Ͳ��ɱ����2���У���ʵ�����ֵ���ϡ�Ǧ�ĵ���ģ�����б�ģ���ֱ�Ϊ16.5��5.8 GPa��ԶС��Mg2Pb��ʵ��������Ѿ��Ʊ���Mg/Pb�Ͻ��俹��ǿ��Ϊ228 MPa��Ӳ��ΪHB157����˵��������Mg2Pb�Ի�����һ����ģ��ǿ�����á�����PUGHԤ�������/���Եľ����о�[25]��G/K��0.5�����ϳ����ԣ���֮��ʴ��ԡ���һ�о��ѱ��㷺Ӧ���ڷ��������仯�����������仯��������Ի����[23]���ɱ�2��֪��Mg2Pb��G/K��0.5��˵�����Ǵ��Ի�������⣬�����Բ��϶��ԣ����ɱȦ�һ��Ϊ1/3�����Բ��ϵIJ��ɱȦͣ�1/3��Mg2Pb������IJ��ɱȦ�С��1/3���ٴ�˵��Mg2Pb��������д��ԡ�
��2 Mg2Pb�������ģ�����б�ģ��������ģ���Ͳ��ɱ�
Table 2 Bulk modulus, shear modulus, elastic modulus and Poisson��s ratio for Mg2Pb

3 ����
1) Mg2Pb��Fermi�ܼ����ϵĵ����ֲ���0~18 eV������������Ϊ-10~-8 eV��-5~0 eV���Լ�-16~-19 eV��-45~-43 eV��Mg��Pb��̬�ܶȵĹ�����Ҫ��Mg��2p�����Pb��5d��������ΪMg��3s�����Pb��6p�����Pb��6s���������С��Mg��2p��Pb��6p�����-5~0 eV�����ص��ϴ��������ڴ���������Ϊǿ�ҵĹ���ӻ���ͬ���أ�Mg2p��Pb 6s��-10~-8 eV��Ҳ��һ������ӻ���
2) ����ܶ�ͼ��������Mgԭ����Χ�д����ĵ�ɴ��ڣ��ʵ��͵Ľ���������������Mg��Pb֮����ڹ��õĵ�ɣ��н�ǿ�������ԣ��Թ��ۼ���ʽ���ڣ��������ɵĻ��䲻�ʹ��ۼ���ռ�������٣���������ռ�����ϴ�Mg2Pb������ʰ�����ԡ�
3) Mg2Pb�ĵ���ģ�����б�ģ���ֱ�Ϊ68.6 GPa��27.9 GPa���ü�������ʵ���������Ǻϣ����������Mg/Pb�Ͻ��ǿ�ȣ�Pugh�����оݺͲ��ɱȾ�����Mg2Pb���д��ԡ�
REFERENCES
[1] BLASKETT D R, BOXALL D. Lead and its alloys[M]. West Sussex: Ellis Horwood Ltd, 1990: 15.
[2] ������, ���ŷ�, �¹���, ����ͤ, ������, �� ��. �����仯����ṹ����. ����: ������ҵ������, 2001: 279.
ZHANG Yong-gang, HAN Ya-fang, CHEN Guo-liang, GUO Jian-ting, WAN Xiao-jing, FENG Di. Structural intermetallics [M]. Beijing: National Defense Industry Press, 2001: 279.
[3] FU C L, PAINTER G S. First principles investigation of hydrogen embrittlement in FeAl[J]. J Mater Res, 1991, 6(4): 719-723.
[4] FOX A G, TABBERNOR M A. The bonding charge density of �¡�NiAl[J]. Acta Metallurgica et Materialia, 1991, 39(4): 669-678.
[5] YOO M H, FU C L. Fundamental aspects of deformation and fracture in high-temperature ordered intermetallics[J]. ISIJ International, 1991, 31(10): 1049-1062.
[6] van DYKE J P, HERMAN F. Relativistic energy-band structure of Mg2Pb[J]. Phys Rev B, 1970, 2(6): 1644-1646.
[7] van ATTEKUM P M TH M, WERTHEIM G K, CRECELIUS G, WERNICK J H. Electronic properties of some CaF2�DStructure intermetallic compounds[J]. Phys Rev B, 1980, 22(8): 3998-4004.
[8] LEE P M. Electronic structure of magnesium silicide and magnesium germanide[J]. Phys Rev, 1964, 135(4A): A1110-A1114.
[9] WOOD D M, Zunger A. Electronic structure of generic semiconductors: Antifluorite silicide and ��-��compounds[J]. Phys Rev B, 1986, 34(6): 4105-4120.
[10] SEGALL M D, LINDAN P J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[11] MARLO M, MILMAN V. Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals[J]. Phys Rev B, 2000, 62(4): 2899-2907.
[12] VANFERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Phys Rev B, 1990, 41(11): 7892-7895.[13] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Phys Rev B, 1976, 13(12): 5188-5192.
[14] PAYNE M C, TETER M P, ALLAN D C, ARIAS T A, JOANNOPOULOS J D. Iterative minimization techniques for Ab-initio total energy calculations: Molecular dynamics and conjugate gradients[J]. Rev Mod Phys, 1992, 64(4): 1045-1097.
[15] RAMACHANDRAN V, IBRAHIM M Md. Third-order elastic constants and the low-temperature limit of the Gr��neisen parameter of Mg2Pb on Axe��s shell model[J]. Journal of Temperature Physics, 1982, 47(3/4): 351-353.
[16] STRINGER G A, HIGGINS R J. Fermi surface of Mg2Pb[J]. Phys Rev B, 1971, 3(2): 506-515.
[17] �� ��, ������, �¹��, �� ��, ������. ��һ��ԭ������XHfO3(X=Ba, Sr)�Ľṹ�����Ժ͵�������[J]. ����ѧ��, 2007, 56(9): 5366-5370.
YU Xiao, LUO Xiao-guang, CHEN Gui-feng, SHEN Jun, LI Yang-xian. First principle calculation of structure, elastic and electronic properties of XHfO3(X=Ba, Sr)[J]. Acta Physica Sinica, 2007, 56(9): 5366-5370.
[18] �� ��, Ҧ����, Ѧ ��. ���Ͽ�ѧ����[M]. ����: ��ѧ��ҵ������, 2006: 58-59.
TAO Jie, YAO Zheng-jun, XUE Feng. Materials science[M]. Beijing: Chemical Industry Press, 2006: 58-59.
[19] WAKABAYASHI N, AHMAD A A Z, SHANKS H R, DANIELSON G C. Lattice dynamics of Mg2Pb at room temperature[J]. Phys Rev B, 1972, 5(6): 2103-2107.
[20] CHUNG P L, DANIELSON G C. ACE report[M]. U.S.: The MIT Press, 1966: 1451.
[21] NYE J F. Physical properties of crystals[M]. Oxford: Oxford University Press, 1985: 28.
[22] ZHANG R F, VEPREK S, ARGON A S. Mechanical and electronic properties of hard rhenium diboride of low elastic compressibility studied by first-principles calculation[J]. Appl Phys Lett,2007:91(20): 201914.1-201914.3
[23] Ҧ ǿ, �� ��, ������, �� ��. TiB2��TiB�������ʵ����ۼ���[J]. �й���ɫ����ѧ��, 2007, 17(8): 1297-1301.
YAO Qiang, XING Hui, MENG Li-jun, SUN Jian. Theoretical calculation of elastic properties of TiB2 and TiB[J]. The Chinese Journal of Nonferrous Metals, 2007, 17(8): 1297-1301.
[24] Ҧ ǿ, �� ��. (Co, Ir)3(Al, W)�������ȶ��Ժ͵������ʵ�һ��ԭ�����о�[J]. �����о���Ӧ��, 2007, 1(4): 281-285.
YAO Qiang, SUN Jian. First-principles study of phase stability and elastic property of (Co, Ir)3( A1, W) precipitate[J]. Materials Research and Application, 2007, 1(4): 281-285.
[25] PUGH S F. Relations between the elastic modulus and the plastic properties of polycrystalline pure metals[J]. Philosophical Magazine, 1954, 45: 823-843.
������Ŀ��������Ȼ��ѧ����������Ŀ(50871049)
�ո����ڣ�2008-10-21�������ڣ�2009-03-14
ͨ�����ߣ��� �£����ڣ��绰��0871-5334093��E-mail: XBYsun@sina.com.cn
[1] BLASKETT D R, BOXALL D. Lead and its alloys[M]. West Sussex: Ellis Horwood Ltd, 1990: 15.
[16] STRINGER G A, HIGGINS R J. Fermi surface of Mg2Pb[J]. Phys Rev B, 1971, 3(2): 506-515.
[20] CHUNG P L, DANIELSON G C. ACE report[M]. U.S.: The MIT Press, 1966: 1451.
[21] NYE J F. Physical properties of crystals[M]. Oxford: Oxford University Press, 1985: 28.
