
DOI��10.19476/j.ysxb.1004.0609.2018.12.20
��������������������Һ�е����������
�� ���������ˣ�����Ӱ���� ���������壬���������� ��
(���ϴ�ѧ ұ���뻷��ѧԺ����ɳ 410083)
ժ Ҫ��
��MnSO4-(NH4)2SO4��Һ�У�������Ϊ��������������̣��ֱ��о������ܶȡ����Һ�ɷ֡��¶ȵ����ض���������Ч�ʡ��ܺĺ����������Ӱ�졣����������ں�Mn2+��ҺŨ��Ϊ30 g/L��(NH4)2SO4Ũ��Ϊ130 g/L��SeO2Ũ��Ϊ0.04g /L����ʼpHֵΪ7.0�������ܶ�Ϊ400 A/m2������¶�Ϊ35 ��ʱ������������������Ч��Ϊ85.8%���ܺ�Ϊ4870.9 kW��h/t���õ��Ľ����̴��ȸ���99.5%������Ϊ��-Mn���봫ͳ���������(0Cr19Ni9)�Աȣ�Mn2+���������������ʼ������λ���ڲ���������������ͬ�����ڵ�������ڿ�������H2���������������ɼ���Mn2+�ĵ������������������Ч�ʣ�����ֱ����ģ�ͬʱ����������������SeO32-�Ļ�ԭ����С���Ӽ�SeO2������������������̵Ĵ��ȡ���ˣ��������߱�����������Ϊ����������̵�DZ����
�ؼ��ʣ�
�̵����������������Ч�����ܺ���SeO2��
���±�ţ�1004-0609(2018)-12-2568-12���� ��ͼ����ţ�TF813���� ���ױ�־�룺A
��������Ϊһ����Ҫ�Ĺ�ҵ�������Ӽ�������Ӧ���ڸ�������ɫ�����Ͻ�͵���Ԫ��������ҵ[1-3]���������������ǵ��͵ġ�����һ�ʡ�(���ܺġ�����Ⱦ����Դ��)��ҵ[4-6]���ڵ�������������У���ĺ���������Ч����Ҫȡ���ڵ缫�����̵ĵ���������ⷴӦ������������ڽ����̵ij�����λ(vs SHE)�ϸ�(��(Mn2+/Mn)=-1.18 V)����Ҫ����������ⷴӦ�Ĺ���λ�������������ʣ����������λ����Һ�ijɷ��Լ��缫���ϵ����ع�ϵ����[7]��Ŀǰ�������о��������̵����������Ӽ��ļ����������̵�Ӱ��[8-15]�������������ϱ������̵ij������̡�������ò�Լ�����Ч��Ӱ����о����١�����̹���һֱ���ò������Ϊ�������ѻ����Ϻ��ϺϽ���Ϊ����Ҳ�й�Ӧ�ñ���[16]����ͬ���������ϻ�Գ�ʼ�̳������������Ӱ�죬ѡ����нϸ��������λ�Ļ����ܽ��ͳ�ʼ�����ⷴӦ���ܺġ���ҵ�ò�����������������λ�Ͻ����̵�Լ��120 mV�����ò���ֳ���������ʱ�����ڵ���������Ч�ʽϵͣ������������Dz���ֱ������Ч����֮��ߡ�Ϊ�˽������ⷴӦ��ɵ��ܺ���ʧ�������������ڲ���ֱ�����κˣ���Ҫ�ڵ��Һ�м���SeO2�����Ӽ����ⲻ�������˳ɱ��������˲�Ʒ��������������˻�����Ⱦ[17]�����Ÿ������̿���Դ�����ѷ�����θ�Ч������������������Դ���ӵ缫���ϽǶȳ��������͵����̵��ܺĶ�����ҵ�Ŀɳ�����չ����Ҫ����[18]��
������̵������������߱��������ܣ��ߵ��������λ��������Һ���ȶ����Ϻõĵ����Ժ���չ�ԡ����������̶Ʋ�Ľ����������[16]�����о�����[19]����MnSO4-(NH4)2SO4��Һ��ϵ�У���ͬ����������Ӧ���������λ�ɸߵ���Ϊ��Al��Mn��Ti��Cu������֡����ڲ�����в�ͬ�̶ȵغ���Ni����Ni����Ͻ��ܴ�������������ⷴӦ�����ⷴӦ��ǿ�ҳ̶��治�����Ni���������Ӷ�����[20-21]��п������̳�ѡ�ô���������������ԭ���ǣ����ĵ�������ǿ����������Һ����ǿ����ʴ�Ժߵ�������� λ[22]���벻�����ȣ������������̵�����������߱������ص㡣
1) ��������/��ԭ��λ(vs SHE)�ϸ�(-1.66 V)��������漫�����Է�����һ�����ܵ�����Ĥ�㣬��Ĥ����������ι̣���������Һ�е���ʴ�����ϲ����ǿ�����Լ�С�������ܽ���̹�������ɵļ�����ʧ�Լ����������������롣2) ���ߵ��������λ����������̵����ڵ���������Ч�ʣ���С���ⷴӦ��ɵ��ܺ���ʧ��3) ����������ϲ�����ᣬ���Լ��Ṥ����Ա�ڳ�װ��ʱ�Ĺ���ǿ�ȣ����⼫�廮�Ƹ�Ĥ��ɵ��ҺpH���������ķ��ա�4) ���ĵ��������ϲ���ֺã�����ߵ����ߵķֲ������ԡ�5) ����ļ۸�ϲ���ְ�ͣ����Խ��ͼ���������ɱ��������ϴ�ľ���������ˣ������нϴ�DZ����Ϊ��������������ϡ�
�������Դ�������Ϊ�о������ص��о������ܶȡ����Һ�ɷ֡��¶ȵ����ض���������Ч�ʡ��ܺĺͳ��������Ӱ�죬�Ż��������������벻��������Աȣ����۴�����Ϊ����̹����������ϵĿ����Ժͼ�������ָ�꣬�ٽ��̵����̵Ľ��ܽ��ġ�
1 ʵ��
1.1 ʵ���Լ�
���Һ�������ƶ�������ˮ�ܽ�һ�������ķ�����һˮ��������(MnSO4��H2O)�������((NH4)2SO4) �Ͷ�������(SeO2) �Ƶá�
1.2 �����ʵ��
�����ʵ�������Ƶ��л��������н��У������ɶԳƷֲ������������ҹ��ɣ�ͨ����Ĥ�ֿ���ÿ���ҵ���Ч���Ϊ0.2 L������Ϊ304����ְ�����壬�����и��20 mm��20 mm��3 mm��������������Cu���ߺ���ݻ�����֬�ܷ��ù������Ϊ4 cm2�Ĺ����缫����������Pb-0.9%Ag�Ͻ�(��������)�������и��15 mm��15 mm��3 mm��������ͬ��������Cu���ߺ���ݻ�����֬�ܷ���2.25 cm2�Ĺ��������
ÿ��ʵ��ȡ��õ���Һ0.4 L��������У�ˮԡ���ȵ��Һ��Ŀ���¶Ⱥ���NH3��H2O��H2SO4��������Һ��pH��Ŀ��ֵ������ĥ����ϴ�õĵ缫�����������ң������������������Ϊ3 cm�����ú���Դ���Ƶ���ǿ�ȣ�ͨ��ˮԡ���ȿ��Ƶ��Һ�¶ȱ仯��Χ�ڡ�1 �棬ʵ����̼�¼�۵�ѹ��������λ�ı仯�����2 h�����ȡ����������ȥ����ˮ��ϴ�缫�������ĵ��Һ���漴����������3%���ظ������Һ�жۻ�����1 min��ȥ����ˮ��ϴ����ٴ��ɡ����������ڵ��ȹķ����������70 ���¸���12 h�������������ǰ�����������ı仯��ͨ��ʽ(1)������������Ч�ʣ�ʽ(2)����������ġ�
 (1)
(1)
ʽ�У���Ϊ��������Ч�ʣ�%�� mΪ�������ǰ�������ı仯��kg��nΪ��������n=2��FΪ�����ڳ�����F=96485 C/mol�� IΪ����ǿ�ȣ�A��tΪ���ʱ�䣬s��MΪ�̵����ԭ��������MMn=55��
 (2)
(2)
ʽ�У�WΪ�ܺģ�kW��h/t��UΪ�۵�ѹ��V��QMnΪ���������������ۺĵ�����975.87 kA��h/t��
����������Ӽ����ϰ��룬����SEM-EDS (Quanta-200) �۲������������ò��ͨ��XRD (D/max-2200) ��2��Ϊ10��~85�㣬ɨ���ٶ�8.0 (��)/min�����¼����������Ľᾧ���ԡ�
����pH3c�;�����ȼƲ�������¼���Һ����ȣ�ȡ��������Һϡ�ͺ���PS-6�͵����ϵ�������ԭ�ӷ�������� (ICP-AES ���� baird) �����Һ��Se��Ũ�ȡ�
1.3 ������������
�����ļ���������PARSTAT 2273�绯ѧ����վ���У����ô�ͳ�����缫��ϵ�������缫ѡ������ʵ����ͬ�������304����ְ壬���������Ϊ1 cm2�������缫��ĥ��ϴ����������Һ(�����Ϊ1:1) �г�����20 min����ˮ�Ҵ�����20 min������ö�������ˮϴ�����κ��ٳ�����20 min���á��Ե缫���ñ������Ϊ4 cm2��ʯī�缫���αȵ缫Ϊ Hg/Hg2SO4/����K2SO4 (0.64 V vs SHE)����������˵�����������е�λ�����ոòαȵ缫��
1.4 �������ԭ��
������̵�������Ӧ��ʽ(3)��(4)��ʾ��
Mn2++2e=Mn�� -1.82 V (3)
-1.82 V (3)
2H2O+2e=H2+2OH-��-1.31 V (4)
ʽ(4)���̳������̵ĸ���Ӧ������ʽ(4)�ķ�Ӧ���ʿ�����Ч�������������Ч�ʡ����ڲ���ֱ�����������������λ�ϸߣ�ʹ��Mn2+��H2O��������ͬʱ�ŵ���������Tafel��ʽ(��ʽ(5))��֪���ڸ�������H2�ڽ����ϵ���������λ������ܶȵĶ���ֵ�������Թ�ϵ������Tafel����a��b��Ҫ�ɵ缫���ϵ��������ʾ�����ͨ������b�ɷ������ⷴӦ���ٿز��衣�����Ի�ƫ������Һ��ʽ(4)ͨ������H2O�ڵ缫����ŵ�����������(��ʽ(6))��������ĸ��Ͻ���(��ʽ(7))��������ĵ绯ѧ����(��ʽ(8))��������[23]��
��=a+b log J (5)
H2O+e=Hads+OH- (b=118 mV/dec, ��=0.5) (6)
2Hads=H2 (b=29.5 mV/dec) (7)
H2O+Hads+e=H2+OH- (b=39.3 mV/dec, ��=0.5) (8)
��������/��ԭ��λ�ϸ����������漫������һ�����ܵ�����Ĥ�㣬���ⷴӦ���̵ĵ������Ӧ��Ҫ������Ĥ���Ͻ��С������ڵ��������ķ�Ӧ����λ��ǰ��Ҫ��Խ����������Ĥ��֮���λ�ݣ�����Ĥ��Ĵ������������ⷴӦ�ı��ۻ�ܣ��Ӷ�������H2�������Ѷȡ����о�����[24]��������Һ�У����������ⷴӦ�����ʿ��Ʋ���Ϊʽ (6)��������������ɷ�Ӧ������������������Ѷ��������������ⷴӦ�ķ������������������Ҫ�ܵ缫���ϵ����ʺ͵��Һ�ijɷֿ��ơ��̵���������Ҫ�����������������Һ�ĵ������������������̵�ˮ�⣬���ڸߵļ�����λ��NH4+Ҳ�ᷢ�����·�Ӧ[7, 13, 16, 25]��
NH4++e��Hads+NH3ads (9)
NH4+�ķŵ�����ӵ缫����������⣬�Ӷ��յ����ⷴӦ����ͨ����������������ɣ��ɼ�������淋Ĵ�����ЧӦ���Ӷ����͵�������ڵķ�Ӧ�ܺġ�
2 ��������
2.1 ��������Ż�ʵ��
���õ��������鷽���������̳��õ�304����������Աȣ������������ڲ�ͬ�����ܶȡ�(NH4)2SO4Ũ�ȡ���ʼMn2+Ũ�ȡ���ʼ����ҺpHֵ����ʼSeO2Ũ�ȡ�����¶�6�������¶Ե���Ч�ʺ��ܺĵ�Ӱ�졣
2.1.1 ���������ܶ�
ʵ���������£����ʱ��2 h����ʼ����ҺpHֵ7.0��(NH4)2SO4Ũ��110 g/L����ʼMn2+Ũ��20 g/L����ʼSeO2Ũ��0.02 g/L������¶�30 �棬�ı����������ܶȣ��������������ܶȶ��̳�����Ӱ�졣ʵ������ͼ1��ʾ��
��ͼ1(a)��֪�������������ܶȵ����ӣ������������ֳ���ͬ�ı仯���ɣ������������ܶ�С��400 A/m2ʱ�����������ܶȵ����������ڵ���Ч�ʵ���ߡ�����������˽���������ͳ��̷�Ӧ֮�⣬�������ų����̵��ܽⷴӦ��ͨ�����������λ���֣�������λ���ŵ����ܶȵ����߶������ƶ���һ����˵��������λ����ʱ���̵����ܽ����ʽϴ���ɵ͵����ܶ�ʱ����Ч�ʵĽ��͡������ܶ�Ϊ400 A/m2ʱ������ֺ��������ĵ���Ч�ʾ��ﵽ���췶Χ�ڵ����ֵ���ֱ�Ϊ82.68%��82.93%��������ֵ�����ܶȺ����Ч�ʿ�ʼ�½�����������������½����ʴ����������ġ�����µ�[26]���÷ֵ������ߵķ���֤ʵ�˸ߵ����ܶ���������ķֵ����ܶȼ������ӣ��������ܶ������H2O�ķŵ���������Mn2+�ķŵ��ܵ����ƣ����ⷴӦ�Ӿ磬������������Ч�ʽ��͡��������ϴ���������λ�����ڸߵĵ����ܶ���Mn2+�ķŵ����ڲ���ֵģ������ͨ������Һ��pHֵ����֤��������ܶȵ����ӣ�����Һ��pHֵ��������������ƣ�����ֺ����������2 h������Һ��pHֵ�ֱ�Ϊ7.40��7.32��˵�������ܶ����ߺ�������������ⷴӦ�����ң��ý����LU��[27]�Ľ���һ�¡�˵���������������ڽϿ��ĵ����ܶȷ�Χ�ڳ��������̣��������̵��IJ���������
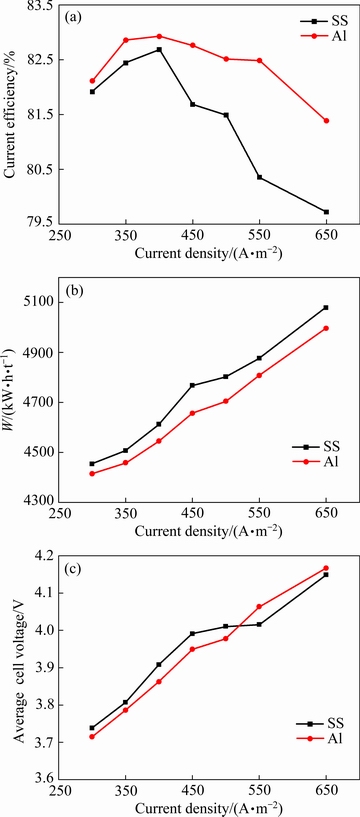
ͼ1 ��ͬ�������������������ܶȶ���������Ч�ʡ��ܺĺ�ƽ����ѹ��Ӱ��
Fig. 1 Effects of cathode current density on cathodic current efficiency(a), energy consumption(b) and average cell voltage(c) with different cathodes
��ͼ1(b)��֪������ܺ�������ܶȵ����ӳ��ֲ������������ƣ���۵�ѹ�ı仯���ƻ���һ�£�˵���ܺ����ӵ�ԭ����Ҫ�����ڲ۵�ѹ�����ߡ�
��ͼ1(c)��֪�������������ܶȴ���450 A/m2ʱ��ƽ���۵�ѹ����һ����С��ƽ̨�������������ܶȳ���500 A/m2����������ƽ����ѹ���Ը��ڲ���������ġ���ѡ������ܶ�ʱ��Ҫͬʱ���������IJ����Ͳ۵�ѹ�������ܶȽϵ�ʱ�۵�ѹ�ϵͣ���λʱ���������IJ���Ҳ�ϵ͡��ۺϿ��ǣ�����ѡ��400 A/m2Ϊ���������ܶȡ�
2.1.2 (NH4)2SO4Ũ��
ʵ���������£����ʱ��2 h�������ܶ�400 A/m2����ʼ����ҺpHֵ7.0����ʼMn2+Ũ��20 g/L����ʼSeO2Ũ��0.02 g/L������¶�30�棬�ı�(NH4)2SO4Ũ�ȣ�����(NH4)2SO4Ũ�ȶ��̳�����Ӱ�졣ʵ������ͼ2��ʾ��
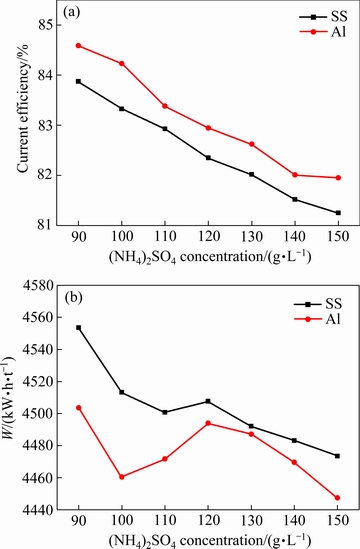
ͼ2 ��ͬ����������(NH4)2SO4Ũ�ȶ���������Ч�ʺ��ܺĵ�Ӱ��
Fig. 2 Effects of cathode current density on cathodic current efficiency(a) and energy consumption(b) with different cathodes
��ͼ2��֪����(NH4)2SO4Ũ�ȵ����ӣ���������Ч�����͡���(NH4)2SO4�ļ�����������Һ�ĵ絼�ʣ���С��Һ���裬������С��������֮�����Һ��ѹ����(NH4)2SO4Ũ����90 g/L����150 g/Lʱ���۵�ѹ��3.9 V����3.7 V��(NH4)2SO4Ũ�ȵ����ӻ����������ӷŵ���̵ĸ����ԣ���ʽ(8)��֪��NH4+�������ŵ��������Һ�е�NH3Ũ�ȣ���Һ�е�Mn2+������NH3�γ�Mn(NH3)2+��Mn(NH3)22+��������λ��[16]���̰�����������õĻ����������Mn2+��Ũ����(NH4)2SO4Ũ�ȵ����Ӷ���С���Ӷ�������Mn2+��ˮ�⣬��ͬʱҲ������Mn2+�ŵ練Ӧ�Ļ�ܡ��̰������ŵ�ͨ���������²��裺1) ��ȥ��λ�壻2) �ŵ��γ�����̬��Mnads+��3) Mnads+�ŵ���뾧���γɽ�����[28]���̰��������γ������˽����̵������Ѷ�[7]�����µ���Ч���������Ũ�ȵ����߶����͡�ͬʱ���缫�����HadsҲ��(NH4)2SO4Ũ�ȵ����Ӷ����ӣ���������������Ч������Hads���ɣ����ڸ�Ũ�ȵ�(NH4)2SO4��Һ�У��������ĵ���Ч�ʸ��ߣ�����ܺĵ��ڲ���������ġ�
���������������ò��֪���͵�(NH4)2SO4Ũ���£������̱���ֲ��н϶�֦������(NH4)2SO4Ũ�ȸ���110 g/L��֦����ʧ����(NH4)2SO4Ũ�ȹ���ʱ��NH4+Ũ�ȵ����ӻ�Ӿ簱�Ļӷ�������̹����л������Եİ�������[29]���Ӷ��������������ϵ�а�����ʧ���ۺϿ��ǣ�����ѡ��(NH4)2SO4Ũ��130 g/L��
2.1.3 ��ʼMn2+Ũ��
ʵ���������£����ʱ��2 h�������ܶ�400 A/m2����ʼ����ҺpHֵ7.0��(NH4)2SO4Ũ��130 g/L����ʼSeO2Ũ��0.02 g/L������¶�30 �棬�ı��ʼMn2+Ũ�ȣ�����Mn2+Ũ�ȶ��̳�����Ӱ�졣ʵ������ͼ3��ʾ��
��ͼ3��֪����������Ч�����ʼMn2+Ũ�ȵ����߶����ӣ��ܺ������ʼMn2+Ũ�ȵ����߶����͡���Һ�е�����Mn2+���ʼMn2+Ũ�ȵ����߶����ӣ���Mn2+Ũ�Ƚϵ�ʱ����������������������ԣ�����Һ��pHֵѸ�����ӣ���Mn2+Ũ�Ƚϸ�ʱ�����ⷴӦ�ܵ�����[12]���Ӷ����ӵ���Ч�ʣ������ܺġ�����Mn2+Ũ�ȸ���30 g/L�缫������OH-������������Mn2+�γ�Mn(OH)2�������˵����̵ĸ����ԡ�ͬʱMn2+Ũ�ȸ���30 g/L��缫�����֦�����������������˳������ƽ���ȡ�
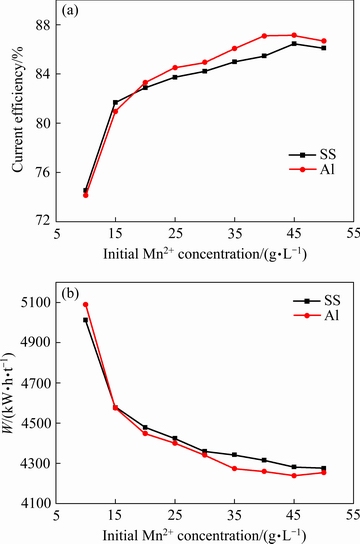
ͼ3 ��ͬ����������Mn2+Ũ�ȶ���������Ч�ʺ��ܺĵ�Ӱ��
Fig. 3 Effects of Mn2+ concentration on cathodic current efficiency(a) and energy consumption(b) with different cathodes
�ԱȲ�ͬ��������֪������ʼMn2+Ũ�ȵ���20 g/Lʱ���ڳ��������̵Ĺ����У�����������ĵ���Ч���Ը����������ġ�����ԭ�����������Mn2+���������ϵķŵ����ʸ����ڲ���������ϵģ���Mn2+�ŵ�Ͽ�ʱ����ʼMn2+Ũ��Խ�ͣ���Խ������ɵ缫����Mn2+��ƶ�����̵ĵ�ᾧ���裬����Ч����֮���͡�����ʼMn2+Ũ�ȸ���20 g/L�鹦�������õ������������������������������̵ĵ���Ч�ʸ��ڲ�����������ۺϿ��ǣ�����ѡ���ʼMn2+Ũ��30 g/L��
2.1.4 ��ʼpHֵ
ʵ���������£����ʱ��2 h�������ܶ�400 A/m2����ʼMn2+Ũ��30 g/L��(NH4)2SO4Ũ��130 g/L����ʼSeO2Ũ��0.02 g/L������¶�30 �棬�ı�����Һ��ʼpHֵ�������ʼpHֵ���̳�����Ӱ�졣ʵ������ͼ4��ʾ��
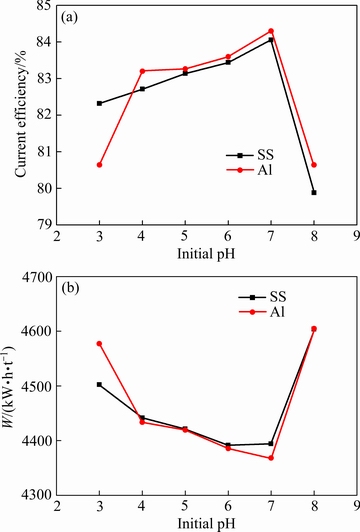
ͼ4 ��ͬ���������³�ʼpHֵ����������Ч�ʺ��ܺĵ�Ӱ��
Fig. 4 Effects of initial pH value on cathodic current efficiency(a) and energy consumption(b) with different cathodes
��ͼ4��֪������Һ��ʼpHֵ�����ߣ������������ֳ����Ƶı仯���ɣ��ڵ͵ij�ʼpHֵʱ������Ч�����ʼpHֵ�����Ӷ����ӡ�����ʼpHֵ������7ʱ�������������������ĵ���Ч�ʷֱ�������84.06%��84.30%���������ӳ�ʼpHֵ��8����Ч���ֱַ���79.88%��80.64%��ʵ����̷��֣��۵�ѹ���������ʼpHֵ�����仯��˵���ܺ���Ҫ����������Ч��Ӱ�졣��ʼpHֵԽ�ͣ��缫��������ⷴӦԽ���ң����ⷴӦ���̳���������ܽⷴӦ�ή�͵���Ч�ʡ���Mn-(NH4)2SO4-H2Oϵ�ĵ�λ-pHͼ[1]��֪������ҺpHֵ����7.5��Mn(OH)2�������ɣ�����Һ����ɫ��Ϊ��ɫ����������Mn(NH3)2+��Mn(NH3)22+��������Ũ����pHֵ�����Ӷ����ӣ����ɵ�Mn2+��pHֵ�����Ӷ����ͣ��Ӷ�������������Ч�ʣ�������Һ�ij�ʼpHֵ���˸���8��
�ԱȲ������������������֪������ʼpHֵ����3ʱ������������ĵ���Ч�ʽϸߣ��ܺĽϵͣ�����ʼpHֵ���ڵ���4���������ĵ���Ч�ʽϸߣ��ܺĽϵ͡���XING��[30]���о���֪����-MnSe���������������������̵��ĵ���Ч�ʡ�������������ȶ�����Ҫ�ܵ�λ����Һ��pHֵ���ƣ��ڵ�pHֵ������MnSe�����ﲻ�ȶ������ܽ����ƴ��ڳ������ƣ���pHֵ����3��MnSe�������������ȶ�����pHֵ����3ʱ����������MnSe����������������ȶ��Խϲ�����˳�ʼ�Ʋ���κ˺�������ʹ�������ĵ���Ч��ƫ�͡�pHֵ���ڻ����4��MnSe�������������ϵij������ƴ����ܽ����ƣ���������������������������ﵽ�ȶ�״̬����ʱ�������ĵ���Ч�ʸ��ڲ���ֵġ��ۺϿ��ǣ�����ѡ���ʼ����ҺpHֵΪ7��
2.1.5 ��ʼSeO2Ũ��
ʵ���������£����ʱ��2 h�������ܶ�400 A/m2����ʼMn2+Ũ��30 g/L��(NH4)2SO4Ũ��130 g/L����ʼ����ҺpHֵ7.0������¶�30 �棬�ı��ʼSeO2Ũ�ȣ�����SeO2Ũ�ȶ��̳�����Ӱ�졣ʵ������ͼ5��ʾ��
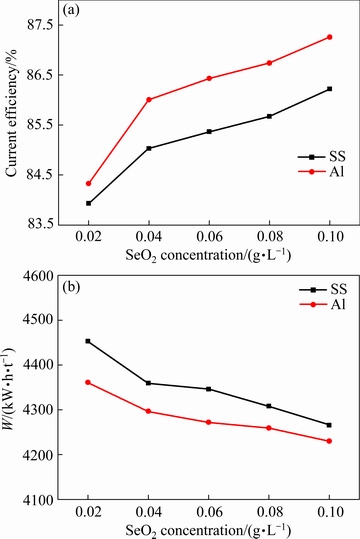
ͼ5 ��ͬ���������� SeO2Ũ�ȶ���������Ч�ʺ��ܺĵ�Ӱ��
Fig. 5 Effects of SeO2 concentration on cathodic current efficiency(a) and energy consumption(b) with different cathodes
��ͼ5 (a) ��֪����SeO2Ũ�ȵ����ӣ���������Ч�ʾ������ߣ�SeO2Ũ�ȳ���0.02 g/L�����Ч���������Ʊ仺����SeO2Ũ����0.02 g/L������0.1 g/Lʱ���������������������Ӧ�ĵ���Ч�ʷֱ���83.93%��84.33%������86.22%��87.26%����ͼ5 (b) ��֪���ܺĵı仯��������������Ч���෴���ܺĵĽ�����Ҫ�����ڵ���Ч�ʵ���ߣ��۵�ѹ�����ȶ���3.80 V���ҡ������о�����[8-16]��SeO2����ǿ���������ⷴӦ�����Ӳ���ֱ�����������λ�����Ե��Һ��SeO2��Ҫ��SeO32-����ʽ���ڣ���������SeO32-��������ԭΪSe0�����������������õ����ӻ�ԭΪ��������ӣ����Ķ�������������ڵ缫����������������������Ѷ�[31]��MnSe��������γɿ��Դٽ�������������ĵ�ᾧ����SeO2�ļ������������ӵ���Ч�ʣ�����������ܺġ�
����ICP-AES������������ڲ�ͬ�����µ��2 h������Һ�е�����Ũ�ȣ������ͼ6��
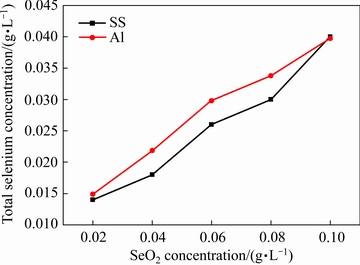
ͼ6 ��ͬ����������SeO2Ũ�ȶԵ���Һ������Ũ�ȵ�Ӱ��
Fig. 6 Effect of SeO2 concentration on total selenium concentration in catholyte after electrolysis with different cathodes
��ͼ6��֪����SeO2Ũ�ȵ����ӣ�SeO2��������Խ��˵���и���������������빲������Ӧ���Ա�����������֪����������������������ʱ������Һ�е�����Ũ�ȸ��ߣ�˵������������������ʱ���ĵ�SeO2���١�ȡ��ʼSeO2Ũ��Ϊ0.04 g/L���Һ�õ��������������SEM-EDS�������������Ͳ������������������������ֱ�Ϊ0.34%��0.72%���̺����ֱ�Ϊ99.66%��99.28%�������������е����������Ե��ڲ�����������ý��������Һ��ICP��������Ǻϣ�˵��������������������̿��Խ������������е�������������SeO2�����ġ�
Ϊ�˶Ա�SeO32-��������������ĵ绯ѧ��Ϊ��ȡ��ʼSeO2Ũ��Ϊ0.1 g/L�ĵ��ҺΪ�о�������-0.8~-1.9�ַ�Χ�ڽ���ѭ������ɨ�裬��Һ�¶�Ϊ30 �棬ɨ���ٶ�Ϊ20 mV/s��������ͼ7��ʾ��

ͼ7 ��ͬ���������µ�ѭ����������
Fig. 7 Cyclic voltammetry with different cathodes
��ͼ7��֪����-0.8~-1.9�ֵ�λ�����ڣ������������ֳ����Բ�ͬ�������绯ѧ��Ϊ����XU��[12]���о��ͺ����CV���߿�֪���õ�λ��������Ҫ����H2��������Ӧ��SeO32-�Ļ�ԭ��Ӧ���ڸ���ɨ������в���������ĵ����ܶ����Ը�����������˵���õ�λ�����ڣ���������������ⷴӦ��Ϊ���ҡ�ͼ7�в�ͼ��ʾΪ-0.8~-1.4�ַ�Χ�ڵľֲ��Ŵ�ͼ�������������-0.8 V����ɨ����-1.4 V�����д����������ԵĻ�ԭ��C1(-0.9 V)��C2(-1.25 V)������C1���ӦSeO32-��ԭΪSe0��C2���ӦSe0��һ����ԭΪSe2-��������ɨ������д����������Ե�������A1(-1.1 V)��A2(-0.9 V)������A1���ӦSe2-����ΪSe0��A2���ӦSe0��һ������ΪSeO32- [9, 12, 15, 29]���벻���������ȣ���������ɨ������еĵ����ź���Ӧ����SeO32-�������������Ϸ���������ԭ��Ӧ��˵������������Ч������SeO32-�Ļ�ԭ���������������ϵĵ�������������κˣ�SeO32-�ڲ���������ϵĻ�ԭ��λ��Mn2+�Ļ�ԭ��λ����Se�Ŀ��ٳ����������ṩ�������ڲ�����������κ˵����ģ��ʵ��Һ�в��ֵ�SeO2��ʧ�ڻ��ױ���ij�ʼ�����㡣������ͨ������SeO32-�Ļ�ԭ�Ӷ�������SeO2����ʧ�������������̵Ĵ��ȡ�������������������и��ߵ��������λ����������Ч�ʷ����������ӡ�
���ڵ��Һ�����������е�Se��������SeO2���������Ӷ����ӣ�SeO2���������ܹ��ߡ�ͬʱ������ʱ����ӳ���SeO32-�ڵ�Ӿ�����»ḻ���ڸ�Ĥ����ɸ�Ĥ����谭���ӵĴ������Ӷ����Ӳ۵�ѹ���ۺϿ��ǣ�����ѡ���ʼSeO2�ļ�����Ϊ0.04 g/L��
2.1.6 ��Һ�¶�
ʵ���������£����ʱ��2 h�������ܶ�400 A/m2����ʼMn2+Ũ��30 g/L��(NH4)2SO4Ũ��130g/L����ʼ����ҺpH 7.0����ʼSeO2Ũ��0.04 g/L���ı���Һ�¶ȣ������¶ȶ��̳�����Ӱ�졣ʵ������ͼ8��ʾ��
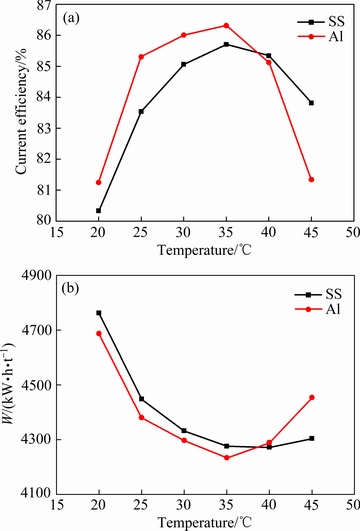
ͼ8 ��ͬ�����������¶ȶ���������Ч�ʺ��ܺĵ�Ӱ��
Fig. 8 Effect of initial temperature on cathodic current efficiency(a) and energy consumption(b) with different cathodes
��ͼ8��֪���¶Ƚϵ�ʱ��������Ч�����¶�����Ѹ�����ӣ����¶ȸ���35 ������ͣ��ܺ������¶ȵ������Ƚ��ͺ����������¶���������Һ�ĵ絼�ʣ��ӿ����ӵĴ��ݺ���ɢ����Ч�ؽ�����Һ����Ͳ۵�ѹ�����Һ�¶���20 ��������45 ��ʱ���������������������Ӧ�IJ۵�ѹ�ֱ���3.92 V��3.90 V������3.71 V��3.70 V��ͬʱ�������¶�����������߷�Ӧ�����ʣ����ͷ�Ӧ�Ļ�ܴӶ������̵ij������������¶ȵ�ͬʱҲ�ή���������������λ���������ⷴӦ�������Ĵ��������ᵼ�³����������ǵ�����ͬʱ�¶ȹ��ߺ���Һ��ϵ��ø�Ϊ���ӣ���Һ�е�Mn2+�ڸ������������ױ�����[32]����ɵ���Ч�ʵĽ��ͺ��ܺĵ����ӡ�
�Ա�����������֪�����¶ȵ���35 ��ʱ���������ĵ���Ч�ʸ��ڲ�������������¶ȸ���35 ����������ĵ���Ч��Ѹ�ٽ��͡�����Ч�ʽ��͵�ԭ���������̳�����������������������������������ϲ�����������ء�˵���������Ը��µ����������ϲ����������������ԭ�������ڸ��������£���������������λ���ͽϿ죬��ɵ缫���淢��ǿ�ҵ����ⷴӦ�������˶Ʋ�����Ľ����������������ڣ���Ƭ�ϱ�������ѹ��С��������ѹ�����ӵ�һ���̶�ʱ����Ƭ�����������ѳɾ������ۺϿ��ǣ�ѡ�����¶�Ϊ35 �档
2.2 ��ѵ������
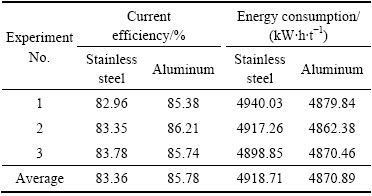
���ݵ���������ʵ�������ó���ѵ���������£������ܶ�400 A/m2��(NH4)2SO4Ũ��130 g/L����ʼMn2+Ũ��30 g/L����ʼpH 7.0��SeO2Ũ��0.04 g/L������¶�35�档Ϊ�����������ʵ��Ч����ȡ��Ч���Ϊ41.25cm2������Ͳ���ְ�����������Ч���Ϊ23.20 cm2��Pb-0.9%Ag�Ͻ��������������3��ƽ����֤ʵ�飬������1��ʾ���ɱ�1��֪�������������µ��2h���벻���������ȣ��������ĵ���Ч����83.4%��ߵ�85.8%���ܺ���4918.7 kW��h/t���͵�С4870.9 kW��h/t�����ֳ��ܺõĽ��ܽ��ĵ�DZ����
��1 �Ż�������ƽ����֤ʵ����
Table 1 Confirmatory experiment results under optimum conditions
2.2.1 ����̵ı�����ò
���Ż������£��������������õ����̵ı�����ò��ͼ9��ʾ����������Ҫ���ֿ�״������״�����ѵ����ɣ��������ֲ���Ϊ���ȡ����У��������������̿���ƽ�����ܣ������ϴ� (10~15 ��m)���������Կ�״Ϊ��������ֱ����������̿�������⻬��������С(5~10 ��m)����������״Ϊ����
2.2.2 ����̵�XRD��
�������Ͳ���ֱ�������XRD����ͼ10��ʾ�����������õ��ĵ�����ﶼ�Ǧ�-Mn�������������ϳ����̵������ǿ�Ȱ�����˳��ݼ���(411)��(721)��(400)��(510)������������ϳ����̵������ǿ�Ȱ�����˳��ݼ���(411)��(721)��(510)����������������̱�¶��(400) �ij���ʹ�������ò�ϲ��������������ͬ��SULCIUS��[33]�ڲ�ͬ�缫�����ϳ���������ʱ���֣������̵ľ�����Ҫ�������������Ӽ���Ӱ�졣�����������滻��ͳ�IJ�������������еĹ��������¿��Բ�����ͬ�ṹ�Ľ����̡�

ͼ9 ����̵�SEM��
Fig. 9 SEM images of electrolytic manganese from different cathodes
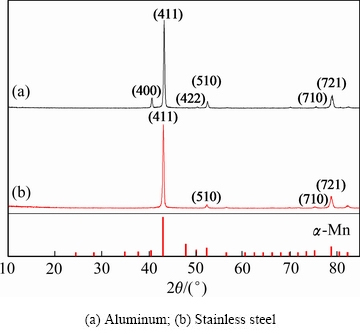
ͼ10 ����̵�XRD��
Fig. 10 XRD patterns of electrolytic manganese from different cathodes
2.3 CV����
Ϊ��һ���������Ͳ������Ϊ����������IJ��죬��-0.8~-2.4 �ַ�Χ�ڶ����缫���Ż����Һ�н�����ѭ������ɨ�裬���Һ�¶�Ϊ35 �棬ɨ������Ϊ5 mV/s��������ͼ11��ʾ��
��ͼ11��֪�������������ֳ����Բ�ͬ�������绯ѧ��Ϊ����������ɨ��-1.7 Vʱ�缫�������������ɣ�˵����ʱ������������������λ��ɨ��-1.98 Vʱ���������ӵĻ�ԭ�壬����-1.3~-1.98 V��Χ����Ҫ�������ⷴӦ�����ij�����Ӧ��-1.98~-2.4 V��Χ����Ҫ�����̵ij�����Ӧ�����ⷴӦ������λ��ɨ��-1.9 Vʱ�����̵������壬��Ӧ����������̵��ܽ⣬�������Ӧ�ķ�ֵ��λ�͵����ܶȷֱ�Ϊ-1.48 V��116.8 mA/cm2�������������ɨ��-1.3 Vʱ�缫��������������ɣ�����λ��ɨ��-1.97 Vʱ���������ӵĻ�ԭ�塣����λ��ɨ��-1.9 Vʱ�����̵������壬���������Ӧ�ķ�ֵ��λ�͵����ܶȷֱ�Ϊ-1.45 V��108.9 mA/cm2���Ա�����������֪���������������λ�ϸ���˵�����нϸߵ��������λ�����������������ⷴӦ��������ʵ��ķ���һ�¡��������Ͳ�������������̵�λ������ͬ��˵���������̹���λ�벻��ֵļ�����ͬ�������̵ij�������û������Ӱ�졣��ͼ11(b)��֪���ڵ���-2.0 V�ĵ�λ�����ڣ���Ȼ�������ĵ����ܶ�С�ڲ���������ģ��������������������������Դ��ڲ���������ģ�˵���õ�λ�����ڣ�����������Ч���������ⷴӦ������Mn2+�ij������Ӷ���Ч��߳�ʼʱ�ڵ���������Ч�ʣ��ý��������ʵ�����Ǻϡ�

ͼ11 ��ͬ���������µ�ѭ����������
Fig. 11 Cyclic voltammetry with different cathodes
3 ����
1) �����������������̵��Ż�����Ϊ�������ܶ�400 A/m2��(NH4)2SO4Ũ��130 g/L����ʼMn2+Ũ��30 g/L����ʼpHֵ7.0��SeO2Ũ��0.04 /L������¶�35 �档�������µ��2 h���Դﵽ86%����������Ч�ʣ�ֱ����Ľϲ������������47 kW��h/t�����ò���Ϊ�������ܡ������ϴ�״�ṹΪ���Ħ�-Mn��
2) ��������������λ���ڲ���֣���ʼ���̵�λ�Ͳ������ͬ���Ӷ���һ�����¶ȷ�Χ�����������������������������Ч�ʡ�ͬʱ����������������SeO32-�Ļ�ԭ����������Se�Ĺ���������ø���Se�����������̡�
3) �봫ͳ�����������ȣ����и��ߵ���������Ч�ʡ����͵�ֱ����ĺ�SeO2���������߱��������ְ���Ϊ�̵������������ϵ�DZ����
REFERENCES
[1] ÷���. �й���ҵ����[M]. ��ɳ: ���ϴ�ѧ������, 2004.
MEI Guang-gui. Technology of China manganese industry[M]. Changsha: Central south University Press, 2004
[2] ������. �й����̿���Դ�͵������̵ķ�չ[J]. �й���ҵ, 2004, 22(3): 26-30.
WANG Yun-min. Resources of Mn-ores and development of EMM[J]. China��s Manganese Industry, 2004, 22(3): 26-30.
[3] ���沨. �й�����������ҵ�ķ�չ����[J]. �й���ҵ, 2014, 32(1): 1-4.
ZENG Xiang-bo. Development trend of EMM in China[J]. China��s Manganese Industry, 2014, 32(1): 1-4.
[4] ����, �ܳ���, ������, �� �[, ������, �� ��. �������ҵ��Ⱦ�������������о�[J]. �������̼���ѧ��, 2013, 3(6): 514-518.
LI Xu-hua, ZHOU Chang-bo, ZHU Ning-fang, WANG Fan, DANG Chun-ge, FANG Gang. Evaluation of the technologies for EMM industry pollution control[J]. Journal of Environmental Engineering Technology, 2013, 3(6): 514-518.
[5] ����, ���Ǹ�, �ε���. �й��������ҵ�������������չ��״�ͷ���[J]. �������̼���ѧ��, 2011, 1(1): 75-81.
DUAN Ning, DAN Zhi-Gang, SONG Dan-na. Current status and directions for development of cleaner production technology of electrolytic manganese metal industry in China[J]. Journal of Environment Engineering Technology, 2013, 3(6): 514-518.
[6] �����, ʱ����, �� ��, �� ��, �����, Ҷ �. ��������������������������ܺĵ�Ӱ��[J]. �й���ɫ����ѧ��, 2016, 26(8): 1774-1781.
WANG Hong-cai, SHI Zhang-ming, LIU Bo, CHEN Bo, YANG Qin-hao, YE Zheng. Influence of material flows in electrolytic manganese metal process on its optimal energy intensity[J]. The Chinese Journal of Nonferrous Metals, 2016, 26(8): 1774-1781.
[7] �。��, �»���, ������, ���¿�. �����̵��ĵ绯ѧԭ������[J]. ��������ѧԺѧ��(��Ȼ��ѧ��), 1997, 17(1): 33-38.
SUN Jian-zhe, CHEN Hu-kui, GUO Jin-bao, YANG Xin-ke. On the electrochemical principles of electrolysis for metal manganese[J]. Journal of Baoji College of Arts and Science (Natural Science) , 1997, 17(1): 33-38.
[8] XUE Jian-rong, ZHONG Hong, WANG Shuai, LI Chang-xin, WU Fang-fang. Influence of sodium silicate on manganese electrodeposition in sulfate solution[J]. Transactions of Nonferrous Metals Society of China, 2016, 26(4): 1126-1137.
[9] JIAO Peng-peng, XU Fu-yuan, LI Jian-hua, DUAN Ning, CHEN Guan-yi, JIANG Lin-hua. The inhibition effect of SeO2 on hydrogen evolution reaction in MnSO4-(NH4)2SO4 solution[J]. International Journal of Hydrogen Energy, 2016, 41(2): 784-791.
[10] PADHY S K, PATNAIK P, TRIPATHY B C, BHATTACHARYA I N. Microstructural aspects of manganese metal during its electrodeposition from sulphate solutions in the presence of quaternary amines[J]. Materials Science and Engineering B, 2015, 193: 83-90.
[11] PADHY S K, PATNAIK P, TRIPATHY B C, GHOSH M K, BHATTACHARYA I N. Electrodeposition of manganese metal from sulphate solutions in the presence of sodium octyl sulphate[J]. Hydrometallurgy, 2016, 165: 73-80.
[12] XU Fu-yuan, DAN Zhi-gang, ZHAO Wei-nan, HAN Gui-mei, SU Ze-hui, XIAO Ke, JIANG Lin-huan, DUAN Ning. Electrochemical analysis of manganese electrodeposition and hydrogen evolution from pure aqueous sulfate electrolytes with addition of SeO2[J]. Journal of Electroanalytical Chemistry, 2015, 741: 149-156.
[13] RUDNIK E. Effect of gluconate ions on electroreduction phenomena during manganese deposition on glassy carbon in acidic chloride and sulfate solutions[J]. Journal of Electroanalytical Chemistry, 2015, 741: 20-31.
[14] DING Li-feng, FAN Xing, DU Jun, LIU Zuo-hua, TAO Chang-yuan. Influence of three N-based auxiliary additives during the electrodeposition of manganese[J]. International Journal of Mineral Processing, 2014, 130: 34-41.
[15] SUN Yan, TIAN Xi-ke, HE Bin-bin, YANG Chao, PI Zhen-bang, WANG Yan-xin, ZHANG Su-xin. Studies of the reduction mechanism of selenium dioxide and its impact on the microstructure of manganese electrodeposit[J]. Electrochimica Acta, 2011, 56(24): 8305-8310.
[16] LU Jian-ming, DREISINGER D, GL��CK T. Manganese electrodeposition��A literature review[J]. Hydrometallurgy, 2014, 141: 105-116.
[17] ����÷, ���Ԫ, ���ֻ�, ���Ǹ�, ��С��, �� ��. �̵����̹ؼ������о������������Է���[J]. �绯ѧ, 2013, 20(3): 282-287.
HAN Gui-mei, XU Fu-yuan, JIANG Lin-hua, DAN Zhi-gang, GAO Xiao-juan, DUAN Ning. Key Factors and techno- economic analyses of manganese electrolysis process[J]. Journal of Electrochemistry, 2013, 20(3): 282-287.
[18] ELSHERIEF A E. A study of the electroleaching of manganese ore[J]. Hydrometallurgy, 2000, 55(3): 311-326.
[19] ILEA P, TISSOT P, ONICIU L. Hydrogen overpotential on several materials for the electrodeposition of manganese from sulfate solutions[J]. Bulletin of Electrochemistry (India), 2000, 16(3): 133-135.
[20] RADHAKRISHNAMURTHY P, SATHYANARAYANA S, REDDY A K N. Kinetics of hydrogen evolution reaction on a stainless steel electrode[J]. Journal of Applied Electrochemistry, 1977, 7(1): 51-55.
[21] OLIVARES-RAMIREZ J M, CAMPOS-CORNELIO M L, GODINEZ J U, BORJA-ARCO E, CASTELLANOS R H. Studies on the hydrogen evolution reaction on different stainless steels[J]. International Journal of Hydrogen Energy, 2007, 32(15): 3170-3173.
[22] �� �, �� ��, ���ҳ�. ��Ӳ���Ͻ���Ϊ������п��������еĵ绯ѧ��Ϊ[J]. ���̿�ѧѧ��, 2016, 38(6): 767-772.
LI Tao, HUANG Hui��GUO Zhong-cheng. Electrochemical behavior of the super high strength aluminum alloy as a cathode in zinc electrodeposition[J]. Chinese Journal of Engineering��2016, 38(6): 767-772.
[23] METIKOS-HUKOVIC M, BABIC R, GURBAC Z, BRINIC S. Inhibition of the hydrogen evolution reaction on aluminium covered by ��spontaneous�� oxide[J]. Journal of Applied Electrochemistry, 1994, 24(4): 325-331.
[24] VIJH A K. Electrolytic hydrogen evolution reaction on aluminum, oxide-covered electrodes[J]. The Journal of Physical Chemistry, 1969, 73(3): 506-513.
[25] KOZIN L F, MASHKOVA N V, MANILEVICH F D. Hydrogen overvoltage at manganese in ammonium-bromide-perchloric solution[J]. Protection of Metals and Physical Chemistry of Surfaces, 2009, 45(1): 25-30.
[26] �����, ����ѧ, �� ��. �����ƶ�п�̺Ͻ�����Ӱ����о�[J]. �����Ϳ��, 2002, 21(1): 1-4.
SHU Yu-de, FU De-xue, MIAO Juan. Studies on the effect of sodium selenate on electrodeposition of zinc-manganese alloy. Electroplating & Finishing[J]. 2002, 21(1): 1-4.
[27] LU Jian-ming, DREISINGER D, GL��CK T. Electrolytic manganese metal production from manganese carbonate precipitate[J]. Hydrometallurgy, 2016, 161: 45-53.
[28] KOZIN L F, MASHKOVA N V, MANILEVICH F D. Kinetics and mechanism of the discharge-ionization of manganese in an ammonium chloride solution[J]. Protection of Metals, 2007, 43(6): 537-541.
[29] ������. ���������������̳�̽[J]. �й���ҵ, 1994, 12(1): 47-49.
WANG Jing-ming. Approach on the cathode process of electrolyte metal manganese[J]. China��s Manganese Industry, 1994, 12(1): 47-49.
[30] FAN Xing, XI Su-yun, SUN Da-gui, LIU Zuo-hua, DU Jun, TAO Chang-yuan. Mn-Se interactions at the cathode interface during the electrolytic-manganese process[J]. Hydrometallurgy, 2012, 127: 24-29.
[31] DHANANJAYAN N, BANERJEE T. Structure of electro- deposited manganese[M]. National Metallurgical Laboratory, 1969.
[32] WEI Qi-feng, REN Xiu-lian, DU Jie, WEI Si-jie, HU Su-rong. Study of the electrodeposition conditions of metallic manganese in an electrolytic membrane reactor[J]. Minerals Engineering, 2010, 23(7): 578-586.
[33] SULCIUS A, GRISKONIS E, KANTMINIENE K, ZMUIDZINAVICIENE N. Influence of different electrolysis parameters on electrodeposition of ��-and ��-Mn from pure electrolytes��A review with special reference to Russian language literature[J]. Hydrometallurgy, 2013, 137: 33-37.
Electrodeposition of Mn from manganese sulfate solution by aluminum cathode
YANG Fan, JIANG Liang-xing, YU Xiao-ying, YANG Jian, LAI Yan-qing, L�� Xiao-jun, LI Jie
(School of Metallurgy and Environment, Central South University, Changsha 410083, China)
Abstract: Aluminum was adopted as cathodic material for manganese electrodeposition from the MnSO4-(NH4)2SO4 solution. The influences of the current density, electrolyte composition and temperature were studied on the cathodic current efficiency, energy consumption and cathodic products. The results show that, the cathodic current efficiency and the energy consumption can reach to 85.8% and 4870.9 kW��h/t, under the following conditions: Mn2+ concentration of 30 g/L, (NH4)2SO4 concentration of 130 g/L, SeO2 concentration of 0.04 g/L, temperature of 35 ��, pH of 7.0 and cathode current density of 400 A/m2. The purity of product can achieve more than 99.5% and the crystal form is ��-Mn. Compared with traditional stainless steel (0Cr19Ni9), the onset potential of manganese electrodeposition on aluminum cathode is not changed, while the hydrogen evolution is suppressed at initial period of electrodeposition. Aluminum cathode can accelerate the discharge rate of Mn2+ and improve the cathodic current efficiency, and then decrease the energy consumption. The reduction of SeO32- on aluminum cathode is weakened, which can reduce the consumption of SeO2 and improve the purity of product. This study indicates that aluminum has the potential to be candidate of stainless steel for manganese electrodeposition.
Key words: manganese electrodeposition; aluminum cathode; current efficiency; energy consumption; SeO2
Foundation item: Project(51374240) supported by the National Natural Science Foundation of China
Received date: 2017-11-01; Accepted date: 2018-02-28
Corresponding author: JIANG Liang-xing; Tel: +86-731-88830474��E-mail: lxjiang@csu.edu.cn
(�༭ ��ѧ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(51374240)
�ո����ڣ�2017-09-05�������ڣ�2017-11-16
ͨ�����ߣ������ˣ������ڣ���ʿ���绰��0731-88830474��E-mail: lxjiang@csu.edu.cn
ժ Ҫ����MnSO4-(NH4)2SO4��Һ�У�������Ϊ��������������̣��ֱ��о������ܶȡ����Һ�ɷ֡��¶ȵ����ض���������Ч�ʡ��ܺĺ����������Ӱ�졣����������ں�Mn2+��ҺŨ��Ϊ30 g/L��(NH4)2SO4Ũ��Ϊ130 g/L��SeO2Ũ��Ϊ0.04g /L����ʼpHֵΪ7.0�������ܶ�Ϊ400 A/m2������¶�Ϊ35 ��ʱ������������������Ч��Ϊ85.8%���ܺ�Ϊ4870.9 kW��h/t���õ��Ľ����̴��ȸ���99.5%������Ϊ��-Mn���봫ͳ���������(0Cr19Ni9)�Աȣ�Mn2+���������������ʼ������λ���ڲ���������������ͬ�����ڵ�������ڿ�������H2���������������ɼ���Mn2+�ĵ������������������Ч�ʣ�����ֱ����ģ�ͬʱ����������������SeO32-�Ļ�ԭ����С���Ӽ�SeO2������������������̵Ĵ��ȡ���ˣ��������߱�����������Ϊ����������̵�DZ����
[1] ÷���. �й���ҵ����[M]. ��ɳ: ���ϴ�ѧ������, 2004.
[2] ������. �й����̿���Դ�͵������̵ķ�չ[J]. �й���ҵ, 2004, 22(3): 26-30.
[3] ���沨. �й�����������ҵ�ķ�չ����[J]. �й���ҵ, 2014, 32(1): 1-4.
ZENG Xiang-bo. Development trend of EMM in China[J]. China��s Manganese Industry, 2014, 32(1): 1-4.
[5] ����, ���Ǹ�, �ε���. �й��������ҵ�������������չ��״�ͷ���[J]. �������̼���ѧ��, 2011, 1(1): 75-81.
[7] �。��, �»���, ������, ���¿�. �����̵��ĵ绯ѧԭ������[J]. ��������ѧԺѧ��(��Ȼ��ѧ��), 1997, 17(1): 33-38.
[22] �� �, �� ��, ���ҳ�. ��Ӳ���Ͻ���Ϊ������п��������еĵ绯ѧ��Ϊ[J]. ���̿�ѧѧ��, 2016, 38(6): 767-772.
[26] �����, ����ѧ, �� ��. �����ƶ�п�̺Ͻ�����Ӱ����о�[J]. �����Ϳ��, 2002, 21(1): 1-4.
[29] ������. ���������������̳�̽[J]. �й���ҵ, 1994, 12(1): 47-49.
