

Electrodeposition behavior of nanocrystalline CoNiFe soft magnetic thin film
LI Jing-feng(���)1,2, ZHANG Zhao(�� ��)2, YIN Jun-ying(����Ӣ)2,
YU Geng-hua(�Ṣ��)2, CAI Chao(�� ��)3, ZHANG Jian-qing(�ż���)2
1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Department of Chemistry, Zhejiang University, Hangzhou 310027, China;
3. Key Laboratory of Energy Resource and Chemical Engineering, Ningxia University, Yinchuan 750021, China
Received 26 September 2005; accepted 8 February 2006
Abstract:
The electroplating behavior of nanocrystalline CoNiFe soft magnetic thin film with high saturation magnetic flux density (Bs��2.1 T) and low coercivity (Hc) was investigated using cyclic voltammetry and chronoamperometry methods in conjunction with the scanning electron microscopy (SEM/EDX). The results show that, under the experimental conditions, the co-deposition of CoNiFe film behaves anomalously due to the atomic radii of iron series elements following the order of rFe��rCo��rNi. In the case of lower electroplating current density, the co-deposition of CoNiFe film follows a 3-D progressive nucleation/growth mechanism, while in the case of higher electroplating current density, which follows a 3-D instantaneous nucleation/growth mechanism. Meanwhile, the change of nucleation mechanism of CoNiFe film with electroplating current density was interpreted theoretically in the light of quantum chemistry.
Key words:
nanocrystalline CoNiFe film; soft magnetism; pulse-reverse electroplating; anomalous co-deposition; electrocrystalliza- tion mechanism;
1 Introduction
Soft magnetic materials are a central component of electromagnetic devices such as step motors, magnetic sensors, transformers and magnetic recording heads. The electrodeposited CoNiFe alloy that possesses a very high saturation magnetic flux density(Bs) and low coercivity (Hc) can meet the requirement for the development of micro-electro-mechanical system(MEMS) and has attracted much attention[1-3]. Generally, the desirable properties of the electrodeposited CoNiFe alloy is directly related to its chemical composition and microstructure[4,5], which mainly depends on the nucleation kinetics and the growth mechanism of the first metallic nuclei formed on the initial substrate[6,7]. Electrocrystallization over different substrates is commonly related to only one kind of nucleation process [8-10]. And complex deposition system consists of two or more nucleation processes with sophisticated transitions between the different types[11].
Characterization of electrocrystallization nucleation/ growth process is usually performed by analyzing the cyclic voltammograms and the current transients obtained using chronoamperometry technique, and a number of different theoretical formalisms have been developed to identify the different nucleation processes, e.g., incorporation of atoms into the nuclei or diffusion and different growth types, e.g., two-dimensional(2D) and three-dimensional(3D)[12-29].
The aim of this study is to obtain the nanocrystalline CoNiFe soft magnetic thin film with high saturation magnetic flux density and low coercivity, especially to probe into the corresponding electrocrystallization mechanism.
2 Experimental
The experimental electrolyte was prepared with AR grade reagents and twice distilled water, and the pH value of the solution was adjusted using H2SO4 and NaOH. CoNiFe deposition was performed using a dual-pulse electroplating workstation (SMD-30, China).
Cyclic voltammetry and chronoamperometry measurements were performed with a commercial Model 660A electrochemical analyzer/workstation (CH Instruments Inc, US). And a three-electrode system consisting of cycloidal polycrystalline brass electrode with an area of 0.502 7 cm2 exposed used as working electrode (WE), saturated calomel (reference) electrode (SCE) and a large bright platinum foil as the counter electrode, was employed. Before each test, the exposed surface of the WE was polished with silicon carbide papers from 3 ��m through 1 to 0.5 ��m, rinsed with the twice distilled water, washed in acetone, rinsed with the twice distilled water again and then dried in air. After the experiments, scanning transmission electron microscopy (SEM, HITACHI S-570, Japan) and material property analysis system (PPMS-9, US) were used to characterize the morphologies and saturation magnetic flux density (Bs, applying the magnetic field in the film plane) of the electrodeposits. Each experiment was repeated at least three runs, and the values reported in this study were the averages of the replicate runs.
The experimental temperature was 25 �� 1 �� con- trolled by thermostat water tank and all potentials were referred to SCE.
3 Results and discussion
3.1 Anomalous codeposition behavior of nanocry- stalline CoNiFe soft magnetic thin film
Table 1 lists the optimized pulse-reverse electro- plating variables and the contents of Fe, Co and Ni in electrodeposited CoNiFe film and the corresponding electroplating solution. It can be seen that, under the experimental conditions, the electroplating of CoNiFe follows the anomalous co-deposition mechanism. So far, many attempts have been made to explain the anomalous co-deposition of iron group alloys, but there is still no universally accepted theory[30].
Generally, it is well accepted that the electro- chemical reduction of iron-group metal ions on the cathode surface obeys the following mechanism[30]:
2H2O+2e��H2+2OH- (1)
M2++OH-��M(OH)+ (2)
M(OH)+��![]() (3)
(3)
![]() +2e��M+OH- (4)
+2e��M+OH- (4)
where M designates iron, cobalt and nickel atoms. Therefore, the reduction rate of M mainly depends on the stability of ![]() or M(OH)+, i.e. the atomic orbital (��A and ��B) overlapping integral (S) of M and O in OH-, the larger the SA-B, the more stable of
or M(OH)+, i.e. the atomic orbital (��A and ��B) overlapping integral (S) of M and O in OH-, the larger the SA-B, the more stable of ![]() or M(OH)+. Based on the electroplating mechanism of iron-group metals represented by Eqns.(1)-(4) and the theory of molecular orbital[31], SA-B is directly proportional to the product of the radial function of the two bonding atoms (Rn, l, A(r)?Rn, l, B(r)).
or M(OH)+. Based on the electroplating mechanism of iron-group metals represented by Eqns.(1)-(4) and the theory of molecular orbital[31], SA-B is directly proportional to the product of the radial function of the two bonding atoms (Rn, l, A(r)?Rn, l, B(r)).
SA-B��Rn, l, A(r)?Rn, l, B(r) (5)
Because the energy of the empty 3d-orbit is less than that of 4s-orbit, the first electron from the cathode will fill into the 3d-orbit preferentially. In this case, the empty orbit of the monovalent iron-base metal ions in ![]() will be the 4s-orbit. For the 4s-orbital of Fe, Co and Ni:
will be the 4s-orbit. For the 4s-orbital of Fe, Co and Ni:
![]() (6)
(6)
therefore, Rn, l, r decreases with the increase of the atomic radius (r) of iron series elements. Because the radii of iron series ions (M2+) follow the following order[32]: rFe��rCo��rNi (Table 2), then SO-Ni��SO-Co��SO-Fe, i.e. the stability of the iron-group metal monohydroxide ions or metal hydroxides can be sorted in the following order: Ni(OH)+��Co(OH)+��Fe(OH)+, which will result in the anomalous codeposition of iron-group metals under the experimental conditions (Table 1). The above stability order of the iron-group metal monohydroxide ions or metal hydroxides is also supported by calculating the crystal field stabilizing energy(CFSE) of Ni(OH)+, Co(OH)+ and Fe(OH)+ using GAUSSIAN94 software [33], on the basis of the electrodeposition mechanism shown in Eqns.(1)-(4) and that, under the electroplating conditions, the OH- is a kind of weak ligands which will result in high spin ligated compounds[34](Table 2).
Certainly, when the electroplating solutions, especially the ligants of the monovalent iron-base metal ions and the electroplating solution pH value are changed, the compound structure of iron-base metal ions will change and normal codeposition will occur[30]. Meanwhile, the change of the parameters of pulse-reverse technique will also change the composition of deposits due to their dissolution in reverse-plating time domain.
Table 1 Electroplating parameters and composition of CoNiFe film

Table 2 Radii of iron-group ions (M2+) [32] and CFSE of Ni(OH)+, Co(OH)+ and Fe(OH)+

3.2 Characterization of nanocrystalline CoNiFe soft magnetic thin film
The morphology and the composition of the optimized electrodeposited CoNiFe film were characterized using SEM and EDX respectively. It can be seen that the surface of the film with the chemical composition of 30.50Fe64.49Co5.01Ni is sufficiently smooth (Fig.1(a)) and possesses nanocrystalline structure (Fig.1(b)). The results indicate that, under the optimized electroplating conditions, the hydrogen generated from Eqn.(1) has less influence on the morphology of the deposit for that, the hydrogen generation will influence the morphology of deposits and usually results in dendrite (or cauliflower shape).
Fig.2 shows the hysteresis curve of the electrodeposited CoNiFe film. It can be seen that the film posses high saturation magnetic flux density (Bs��2.1 T) and low coercivity(Hc), and can meet the requirement for the development of micro-electro-mechanical system (MEMS)[1-3].

Fig.1 SEM images of CoNiFe film: (a) Macrocosm; (b) Enlarged diagram of Fig.1(a)
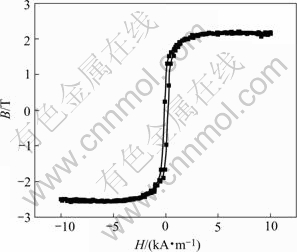
Fig.2 Hysteresis loop of nanocrystalline CoNiFe film with composition of 30.50Fe64.49Co5.01Ni
3.3 Electrocrystallization nucleation and growth mechanism
The electrocrystallization behavior of the optimized nanocrystalline CoNiFe soft magnetic thin film was first investigated using cyclic voltammetry at a sweep rate of 10 mV/s. The sweep potential range is from open circuit potential to -1.6 V and always initiates in the negative direction (Fig.3). It can be seen that there exist nucleation/growth loops in the cathodic branch of the voltammogram, which indicates that the co-deposition of CoNiFe film follows the three-dimensional (3-D) nucleation/grain growth mechanism[35]. In order to characterize the electrocrystallization process in more detail, the initial electrodeposition process of CoNiFe film was investigated using chronoamperometric analysis technique based on the three-dimensional (3-D) nucleation/grain growth mechanism obtained from cyclic voltammetry results.
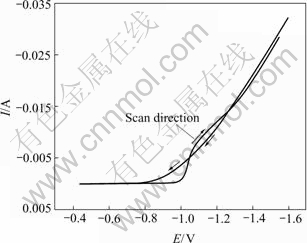
Fig.3 Cyclic voltammograms of brass in CoNiFe electroplating solution
In chronoamperometry experiments, the potential was stepped from the open-circuit potential (about -0.425 V) where no deposition of metal was detected in the cyclic voltammogram, to the potential at which the deposition of CoNiFe would occur. Fig.4 shows the experimental current��time transient curves. It can be seen that, each of the I��t curves consists of an initial spike (within the first 0.02 s) due to the charging of the electrochemical double layer, a subsequent rising portion due to the nucleation process and a posterior decreasing portion due to the diffusion process. The rising section appears to reach its maximum at increasingly shorter time with more negative overpotential (��) steps. The maximum in the current transient at high overpotentials and short time corresponds to the maximum surface area, i.e. the point at which hemi-spherical nuclei are on the point of collision. Meanwhile, when compared the I��t curves at the deposition potential of -1.02 V and -1.06 V, it can be seen that the time for the appearance of the current maximum at -1.02 V is much shorter than that at -1.06 V. The reason may be that the electroplating process of CoNiFe may be controlled mainly by electrochemical reaction at much small �� and controlled by diffusion at high ��, respectively.
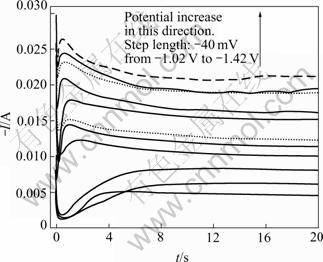
Fig.4 Potentiostatic I��t transients for nucleation of CoNiFe film
Generally, nuclei are preferentially formed on the surface inhomogeneities like emergence points of edge and screw dislocations, atomic disorder, kink sites, or monoatomic steps, etc. And there are a number of literatures describing the electrocrystallization process mathematically[12-29]. However, to date, the most widely employed theoretical model for electrochemical nucleation is the one developed by HILLS et al[11-14, 36]. Even though this theoretical model was based on the nucleation/growth of single metal, it has been widely used to analyze the nucleation/growth of alloys [5, 37].In this model[11-14], the authors described the kinetics of electrolytic phase formation at early stages when diffusion of the electroactive species from bulk solution to the interface was the rate determining step, and the growth of nuclei was considered to be 3D taking into account overlap of diffusion zones, and AFSHR et al[37] and YANG et al[38] believed that the deposition of iron series metal and iron-based alloys tallied this model best. According to this model, the rising portion of the current transient can be described, respectively for the instantaneous nucleation and progressive nucleation by
I(instantaneous)![]() (7)
(7)
![]() (8)
(8)
![]() (9)
(9)
![]() (10)
(10)
where zF is the molar charge of electrodepositing species; D is the diffusion coefficient; c is the bulk concentration of the zinc species; N is the number of nuclei; N�� and AN�� are the total number of active sites for instantaneous nucleation and progressive nucleation respectively; M is the molar mass; ��is the density of the deposited material; and k and k�� are the numerical constants determined by the experimental conditions. Determination of the nucleation process involved was achieved by analyzing the rising section of the current transient and then comparing the curve to the dimensionless theoretical curves obtained from Eqns.(7) and (9), respectively.
For the reason that the optimized nanocrystalline CoNiFe soft magnetic thin film (Fig.1) is obtained using higher initial cathodic current than -0.02 A, only the I��t curves at high ��(Fig.4) were analyzed. Fig.5 shows the relationship of the non-dimensional variables between (t/ tm) and (I/Im)2, where Imax and tmax are the current transient maximum values. It can be seen that the experimental I��t curves closely follow the theoretic progressive or instantaneous nucleation curve in the low and high overpotential domain respectively, and the nucleation rate increases with the increasing ��.
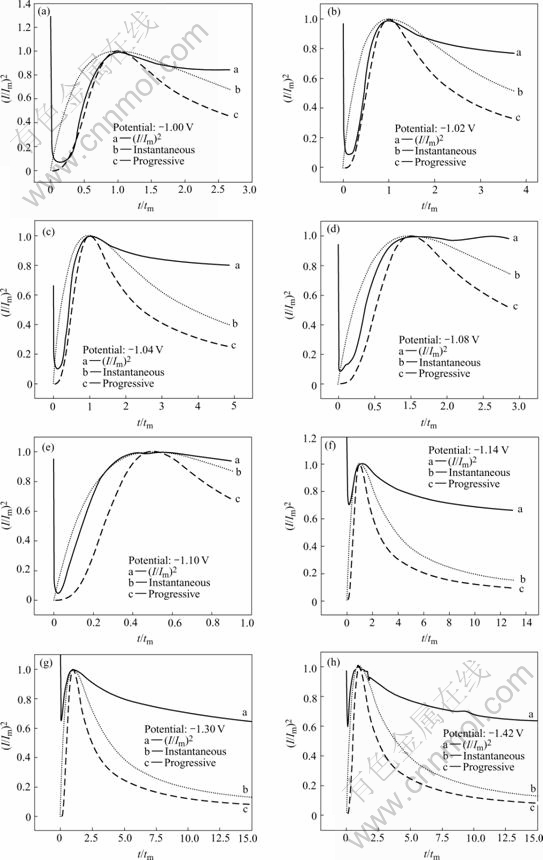
Fig.5 Non-dimensional I/Imax vs t/tmax plot for electrodeposition of CoNiFe film
According to our previous study[39], when the absolute temperature(T) is constant, the electron number (N) in elemental volume(V) of the cathodic surface layer, whose energy is in the gap between ��k and ��k+d��k, is dependent only on the value of the cathode Fermi level (EF).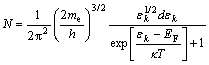 (11)
(11)
where me is the electron mass; h is the Plank��s constant; ��is the Boltzmann constant, ��k is the orbit energy level of the active positive particles, and ![]() . Meanwhile, the Fermi level (EF) of the cathode can be estimated on the bases of quantum chemistry as follows:
. Meanwhile, the Fermi level (EF) of the cathode can be estimated on the bases of quantum chemistry as follows:
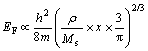 (12)
(12)
where ��, Ms and x are the density, the molar mass and the normal ion value of the cathode metal respectively. Because the ��/Ms of CoNiFe alloy is much larger than that of the original polycrystalline brass substrate (Table 3), which results in that the electron number (N) in elemental volume of the cathodic brass surface layer, whose energy is in the gap between ![]() and
and ![]() , is much less than that of CoNiFe alloy. Therefore, when the overpotential step in chronoamperometry experiments is much lower, i.e. the electroplating current density (J) is sufficiently low, it is difficult for iron group metal ions to obtain electrons from the brass cathode, which will result in the fast growth of nuclei formed previously on the cathode surface due to the high current density distributed on these points, and results in progressive nucleation. In this case, only few granular CoNiFe particles can be observed on the cathode surface after the short-time chronoamperometry experiments.
, is much less than that of CoNiFe alloy. Therefore, when the overpotential step in chronoamperometry experiments is much lower, i.e. the electroplating current density (J) is sufficiently low, it is difficult for iron group metal ions to obtain electrons from the brass cathode, which will result in the fast growth of nuclei formed previously on the cathode surface due to the high current density distributed on these points, and results in progressive nucleation. In this case, only few granular CoNiFe particles can be observed on the cathode surface after the short-time chronoamperometry experiments.
Table 3 Density and molar mass of iron-group elements and copper[32]

On the other hand, when the potential steps to much more negative value in chronoamperometry experiments, i.e. the electroplating current density (J) is much high, the iron group metal ions can more easily obtain electrons from cathode due to the synergism of that the CoNiFe film formed previously possesses higher EF than brass substrate and ![]() which will result in instantaneous nucleation.
which will result in instantaneous nucleation.
It should be mentioned that, when performing chronoamperometric analysis, some authors suggested using the modified I��t curves, which were obtained by subtracting the I��t curves obtained in blank solution (without the electrodeposited metal ions) from those obtained in the electroplating solution (with the electrodeposited metal ions) under exactly the same experimental conditions[40-42]. Generally, this method could eliminate the contribution of currents that are not directly related to metal charging, nucleation and growth phenomena, and should be recommended. However, under the experimental conditions as in this study, the nucleation and growth phenomena of CoNiFe alloys are
always coupled with the evolution of hydrogen as shown in Eqns.(1)-(4), and the hydrogen reduction rate in blank solution is much different from that in electroplating solution (with the electrodeposited metal ions). Consequently, it should be more rational to use the I��t curves obtained in electroplating solution but the modified I��t curves to perform chronoamperometric analysis.
It can also be seen from Fig.5 that, after the current maximum, the experimental (I/Im)2 is much larger than that calculated from the theoretical model. The reason may be that the stability of the iron-group metal monohydroxide ions or metal hydroxides can be sorted in the following order: Ni(OH)+��Co(OH)+��Fe(OH)+ as elucidated above, consequently the content of nickel in the electrodeposited film is much lower than that in the electroplating solution, the concentration of Ni2+ in the vicinity of the cathode should maintain at a relatively steady value (Table 1) during the whole electroplating process. Meanwhile, the nucleation/growth process of CoNiFe deposit is always coupled with the evolution of hydrogen, which can be seen from Eqns.(1)-(4). Therefore, the experimental (I/Im)2 should be much larger than that calculated from the theoretical model, while the decrease of current after the current maximum in I��t curves may originate from the sparsity of Co2+ especially Fe2+ on the cathode surface.
4 Conclusions
1) Nanocrystalline CoNiFe soft magnetic thin film with high saturation magnetic flux density and low coercivity is obtained using pulse-reverse electroplating technique.
2) Under the experimental conditions, the co-deposition of CoNiFe film behaves anomalously. The anomalous co-deposition of CoNiFe film arises because the atomic radii of iron series elements follow the following order: rFe��rCo��rNi, which will result in that the stability of the iron-group metal monohydroxide ions or metal hydroxides can be sorted in the following order: Ni(OH)+��Co(OH)+��Fe(OH)+.
3) In the case of lower electroplating current density, the co-deposition of CoNiFe film follows a 3-D progressive nucleation/growth mechanism. In the case of higher electroplating current density, the co-deposition follows a 3-D instantaneous nucleation/growth mecha- nism.
References
[1] OSAKA T, TAKAI M, HAYASHI K, OHASHI K, SAITO M, YAMADA K. A soft magnetic CoNiFe film with high saturation magnetic flux density and low coercivity [J]. Nature, 1998, 392: 796-798.
[2] BAI A, HU C C. Effects of electroplating variables on the composition and morphology of nickel-cobalt deposits plated through means of cyclic voltammetry [J]. Electrochim Acta, 2002, 47: 3447-3456.
[3] OSAKA T. Introduction of electrochemical microsystem technologies (EMT) from ultra-high-density magnetic recording [J]. Electrochim Acta, 2001, 47: 23-28.
[4] NAKANISHI T, OZAKI M, NAM H S, YOKOSHIMA T, OSAKA T. Pulsed electrodeposition of nanocrystalline CoNiFe soft magnetic thin films [J]. J Electrochem Soc, 2001, 148(9): C627-C631.
[5] PETERSSON I, AHLBERG E. Kinetics of the electrodeposition of Pb circle divide Sn alloys (Part I): at glassy carbon electrodes [J]. J Electroanal Chem, 2000, 485: 166-177.
[6] MARGARITA M H, MANUEL P P, NIKOLA B, IGNACIO G. Identification of different silver nucleation processes on vitreous carbon surfaces from an ammonia electrolytic bath [J]. J Electroanal Chem, 1998, 443: 81-93.
[7] MARGARITA M H, MANUEL P P, NIKOLA B, IGNACIO G. Detailed characterization of potentiostatic current transients with 2D-2D and 2D-3D nucleation transitions [J]. Surf Sci, 1998, 399: 80-95.
[8] HOLZLE M H, RETTER U, KOLB D M. The kinetics of structural changes in Cu adlayers on Au (111) [J]. J Electroanal Chem, 1994, 371: 101-109.
[9] ENRIQUE B, MANUEL P P, NIKOLA B, IGNACIO G. Formation mechanisms and characterization of black and white cobalt electrodeposition onto stainless steel [J]. J Electrochem Soc, 2000, 147(5): 1787-1796.
[10] MANUEL P P, MA T R, IGNACIO G. New insights into evaluation of kinetic parameters for potentiostatic metal deposition with underpotential and overpotential deposition processes [J]. J Electrochem Soc, 1996, 143(5): 1551-1546.
[11] GUNAWARDENA G, HILLS G, MONTENEGRO I, SCHARIFKER B. Electrochemical nucleation (Part ��): General considerations [J]. J Electroanal Chem, 1982, 138: 225-239.
[12] HILLS G J, SCHIFFRIN D J, THOMPSON J. Electrochemical nucleation from molten salts (��): Diffusion controlled electrodeposition of silver from alkali molten nitrates [J]. Electrochim Acta, 1974, 19: 657-670.
[13] GUNAWARDENA G, HILLS G, MONTENEGRO I, SCHARIFKER B. Electrochemical nucleation (Part ��): The electrodeposition of mercury on vitreous carbon [J]. J Electroanal Chem, 1982, 138: 255-271.
[14] SCHARIFKER B, HILLS G. Theoretical and experimental studies of multiple nucleation [J]. Electrochim Acta, 1981, 28: 879-889.
[15] SCHARIFKER B, MOSTANY J. Three-dimensional nucleation with diffusion controlled growth (Part��): Number density of active sites and nucleation rates per site [J]. J Electroanal Chem, 1984, 177: 13-23.
[16] DEUTSSCHER R L, FLETCHER S. Nucleation on active sites (Part IV): Invention of an electronic method of counting the number of crystals as a function of time; and the discovery of nucleation rate dispersion [J]. J Electroanal Chem, 1998, 239: 17-54.
[17] DEUTSSCHER R L, FLETCHER S. Nucleation on active sites (Part V): The theory of nucleation rate dispersion [J]. J Electroanal Chem, 1990, 277: 1-18.
[18] FLETCHER S, HALLIDAY C S, GATES D, WESTCOTT M, WIN T L, NELSON G. The response of some nucleation/growth processes to triangular scans of potential [J]. J Electroanal Chem, 1983, 159: 267-285.
[19] FLETCHER S. Nucleation on active sites (Part II): Prohabilistic formulae for electrical currents [J]. J Electroanal Chem, 1984, 164: 11-16.
[20] DEUTSSCHER R L, FLETCHER S. Nucleation on active sites (Part I): Probabilistic formulae for the numbers of crystals [J]. J Electroanal Chem, 1984, 164: 1-9.
[21] TADANNO A, ASANUMA M, AOGAKI R. Electrochemical nucleation induced by nonequilibrium fluctuations [J].J Crystal Growth, 1996, 166: 1111-1115.
[22] ABYANEH M Y. Extracting nucleation rates from current-time transients (Part I): The choice of growth models [J].J Electroanal Chem, 2002, 530: 82-88.
[23] ABYANEH M Y, FLEISCHMANN M. Extracting nucleation rates from current�Ctime transients (Part II): Comparing the computer-fit and pre-pulse method [J].J Electroanal Chem, 2002, 530: 89-95.
[24] ABYANEH M Y. Extracting nucleation rates from current-time transients (Part III): Nucleation kinetics following the application of a pre-pulse [J].J Electroanal Chem, 2002, 530: 96-104.
[25] AVRAMI M. Kinetics of phase change (I): General theory [J]. J Chem Phys, 1939, 7: 1103-1112.
[26] AVRAMI M. Kinetics of phase change (II): Transformation-time relations for distribution of nuclei [J]. J Chem Phys, 1940, 8: 212-224.
[27] AVRAMI M. Kinetics of phase change (III): Granulation and phase change and microstructure [J]. J Chem Phys, 1941, 9: 177-184.
[28] AOGAKI R. Instability of nonequilibrium fluctuation in electro- chemical nucleation (I): Occurrence of instability [J]. J Chem Phys, 1995, 103(9): 8602-8615.
[29] MANUEL P P, IGNACIO G, NIKOLA B. New insights into evaluation of kinetic parameters for potentiostic metal deposition with underpotential and overpotential deposition process [J]. J Chem Phys B, 2000, 104: 3545-3555.
[30] TSAY P, HU C C. Non-anomalous codeposition of iron-nickel alloys using pulse-reverse electroplating through means of experimental strategies [J]. J Electrochem Soc, 2002, 149: C492-C497.
[31] ZHANG Z, ZHANG J Q, LENG W H, CAO C N. Cooperation behavior of iron and phosphorus in electrodeposition of zinc-iron-phosphorus coatings [J]. Mater Chem Phys, 2002, 77: 497-500.
[32] LIOU C K. Inorganic Chemistry (vol.��) [M]. Changsha: Central South University of Technology Press, 1994. 182.
[33] FRISCH M J, TRUCKS G W, SCHLEGEL H B. et al. Gaussian 94 (Revision E.1) [M]. Pittsburgh PA: Gaussian Inc, 1995.
[34] JIANG Y S. The Structure Chemistry [M]. Beijing: High Education Press, 1997. 197-202.
[35] JAYA S, PRASADA R T, PRABHAKARA R G. Electrochemical phase formation (I): The electrodeposition of copper on glassy carbon [J].Electrochim Acta, 1986, 31(3): 343-348.
[36] SCHINDLER W, HUGELMANN P, HUGELMANN M, KARTNER F X. Localized electrochemical nucleation and growth of low-dimensional metal structures [J].J Electroanal Chem, 2002, 522: 49-57.
[37] AFSHR A, DOLATI A G, GHORBANI M. Electrochemical characterization of the Ni-Fe alloy electrodeposition from chloride-citrate-glycolic acid solution [J]. Mater Chem Phys, 2002, 77: 352-358.
[38] YANG Z N, ZHANG Z, ZHANG J Q. Electrodeposition of decorative and protective Zn-Fe coating onto low-carbon steel substrate [J]. Surf Coat Technol, 2006, 200(16-17): 4810-4815.
[39] ZHANG Z, LENG W H, SHAO H B, ZHANG J Q, WANG J M, CAO C N. Study the behavior of Zn-Fe alloy electroplating [J]. J Electroanal Chem, 2001, 516: 127-130.
[40] STOYCHEV D, PAPOUTSIS A, KELAIDOPOULOU A, KOKKINIDIS G, MILCHEV A. Electrodeposition of platinum on metallic and nonmetallic substrates��selection of experimental conditions [J].Mater Chem Phys, 2001, 72: 360-365.
[41] KELAIDOPOULOU A, KOKKINIDIS G, MILCHEV A. Nucleation and growth of metal catalysts (Part I): Electrodeposition of platinum on tungsten [J].J Electroanal Chem, 1998, 444: 195-201.
[42] MILCHEV A, STOYCHEV D, LAZAROV V, PAPOUTSIS A, KOKKINIDIS G. Electrocrystallisation of metal catalysts: nucleation and growth of platinum on a titanium electrode [J].Journal of Crystal Growth, 2001, 226(1): 138-147.
Corresponding author: LI Jin-feng; Tel: +86-731-8830270; E-mail: lijinfeng@mail.csu.edu.cn
Abstract: The electroplating behavior of nanocrystalline CoNiFe soft magnetic thin film with high saturation magnetic flux density (Bs��2.1 T) and low coercivity (Hc) was investigated using cyclic voltammetry and chronoamperometry methods in conjunction with the scanning electron microscopy (SEM/EDX). The results show that, under the experimental conditions, the co-deposition of CoNiFe film behaves anomalously due to the atomic radii of iron series elements following the order of rFe��rCo��rNi. In the case of lower electroplating current density, the co-deposition of CoNiFe film follows a 3-D progressive nucleation/growth mechanism, while in the case of higher electroplating current density, which follows a 3-D instantaneous nucleation/growth mechanism. Meanwhile, the change of nucleation mechanism of CoNiFe film with electroplating current density was interpreted theoretically in the light of quantum chemistry.
