
���±�ţ�1004-0609(2016)-09-1967-09
�Ͻ�Ԫ��Zr������Ϳ��ؼ������ǿ��ЧӦ������
����ǿ1, 2���� ��1, 2, 3, 4���� ��3, 4
(1. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ����ɳ 410083��
2. ���ϴ�ѧ ��������ɫ���������ص�ʵ���ң���ɳ 410083��
3. ���ϴ�ѧ ��ĩұ������ص�ʵ���ң���ɳ 410083��
4. ���ϴ�ѧ �����о�Ժ������ 518057)
ժ Ҫ��
��Ը�������Ϳ���йؼ��Ĺ��ɲ�/����������棬ͨ����һ��ԭ����������ѧ��ģ���ܶȷ������㣬�������Ԫ�ض�g-Ni(Al)/��-Al2O3����ṹ��ƫ�����ͽ����ϵ�Ӱ�죬�Լ��ڻ����ж�����(��)�Ķ����������ʾ��ﯾ�����ͬ��Ԫ�������ƵĽ���ǿ�����ã���ǿ���������Թ���Ϊ3�֣����ڻ�������Ч����������������к�����ƫ�����ڽ��洦�û����Լ���ƫ����Խ����Σ���Լ��ڽ���ƫ����ֱ��ǿ�������ϣ���ʹ���������뻯ѧ�����Ƚ���ķ��빦���3����
�ؼ��ʣ�
����Ϳ��������ǿ�����Ͻ��������һ��ԭ����
��ͼ����ţ�TG132.32���� ���ױ�־�룺A
��20����60��������������Ͻ����Ի���ϵ�Ͻ���гɷָ��죬��Ϊ�������ºϽ�����з�����Ҫ;��֮һ���Ͻ�ͨ����ָ��ԭ�����Ͻ�Ԫ�صĻ����ϣ�ͨ������һ�ֻ�������Ľ�����ǽ���Ԫ�أ����ǡ���ı��κ��ȴ����������Ը��ƺϽ����ѧ���ܡ���ʴ�ԡ������Եȡ���Ԫ�صļ�������Ͻ���ϵ���죬��ǵ��ʽṹ�ּ�����һ��С��0.1%(Ħ����������ͬ)�����ȸֺͲ���ּ�����ԼΪ0.5%�����Ͻ������һ��С��1%�����ºϽ������Ϊ1%~2%����Ȼ��������Ͻ����û���������ȷ�����Ͻ����Ѿ��㷺��Ӧ���ڸ����ºϽ���ϵ���ơ��Ʊ���������ʵ���С������Ͻ�Ԫ�صļ��������٣�һ������ƶ��乤������Ӧ�����ǿ����ϵ������Ҫ���κ˺ͳ����Լ���λ�����ǽṹ�������ȷ�����������йء�
�����Ͻ��������Ϳ��(Thermal barrier coatings, TBCs)�ں��շ�������ȼ���ֻ������¹���ȼ�ϵ�ص��ڶ���Ҫ����ϵͳ�з����Źؼ������á���Ϳ���Ʊ�����۹����У����������ɲ�Ͻ�(Bond coat, BC)�������γ���������100 ��m���ҵ���������(Thermally-grown oxide, TGO)�������������ÿ���Ч��������Ӧ������Ϊ����Ͻ��ṩһ���Ŀ���������������������ʵ��ͷ��۾���֤�����������γɵ�BC/TGO���������������ճ�����������������Ϳ��(YSZ)��ֱ�Ӱ��䡣������֮��BC/TGO������ǿ�ȼ����˻��ݱ䣬�Ǿ�����������Ϳ��ɿ��Ժͷ��������Ĺؼ���EVANS��[1]��GLEESON [2]�Լ����ڽ�������[3]�Ⱥ����о�������ʽ����BC/TGO����ʧЧ���Ƽ���ӦBC�Ͻ��Ż���ƣ����й��꾡�ķ������ܽᡣ����ʵ���о�������BC�Ͻ��в����Ļ��ȼ�����������Ҫ����Ԫ��(��S)������BC/TGO�����Ϸ���ǿ��ƫ�ۣ������������������ϣ�������������Ȳ���ȫʧЧ����Ҫ����[4-6]����BC�Ͻ���������ϡ�����ϡ��Ԫ��(�� Hf��Y��Ce��Zr��)��������������ɵõ���ͬ�̶ȵػ���[7-12]����Щ�Ͻ�Ԫ�ص����û��ƣ�һ�������ֻ������裺1) ���ںϽ��������Ч��������S���Ӷ����ͺ����������к�ƫ����2) �ڽ������ɼ���ֱ��ǿ�������ϡ���Щ������Ժܺõؽ����ڶ��ʵ��۲죬����δ���ֱ�ӵ�ʵ����֤[9]����ʵ�ʷ��۹��������£���Щ���ƿ��ܵ�����ͬʱ�������ã����Ӱ�졣��������ʵ�鷽���ͼ�����Ŀǰ�����Գ�ֽ�ʾ�Ͻ�Ԫ�صĸ��ӽ���ЧӦ��
����Ϳ������о�����������Щ���ѣ��ǵ�ǰ�����ѧ�еĹ������⣬�������Ԥ��һ�������������ǿ�ȣ����ȷ������ǿ�ȵ���ҪӰ�������Լ������ؼ�����ϵ���Ƿ���ڳ����������ǿ�ȵĿ��ܣ��Լ���λ��ڽ��濪չ��������Ż��ȡ��ش���Щ���⣬�������ȳ����ʶ����ṹ�ͽ���ǿ�����Ʊ�����۹����������Լ��Ͻ��ڲ���ѧ�ɷֵ�����ԡ���־�ֵ�[13]���þ���������ۺ���ķ˹-����-������-��(Thomas-Fermi-Dirac-Cheng, TFDC)���ۣ�ͨ������۵����ܶ�����������Ӽ��㣬���������Ͻ�Ԫ��Crƫ�����մɾ��Ȳ�(YSZ)/BC�Ͻ�����ϵ�Ӱ�졣Ȼ��������������ۺ�TFDC�����о�������ϵ��ȷ�Էdz����ޣ�����YSZ��BC�Ͻ�֮��ʵ���ϻ�������TGO�㣬BC/TGO�������Ŀǰ����Ϊ��������Ϳ��ϵͳ����Σ�յĽ��档����������һ��ԭ�����㷽�����첢�ɹ�Ӧ���ڽ����о���Ϊ��ʾ��������ԵĽ�����������ṩ���µ��о��ֶΡ�CARLING[14]��JARVIS��[15]�Ⱥ�չ���BC/TGO����ĵ�һ��ԭ�������о�������Ԥ�ⲻͬ�Ͻ�Ԫ�ضԽ����ϵ�ǿ����Ӱ�졣����Щ�о�δ���Ǹ�Ԫ���ڽ����ƫ�������;����ԣ�Ҳδ���Ǹ�Ԫ�ض�����S�Ŀ��ܶ������ر��Ǻ�����TGO���������ѧ�����ص㣬��ȫ�����ǺϽ�Ԫ�ػ�Ⱥ��¶ȵ�Ӱ�죬�Ӷ������˽����۽ṹ������ѧ��������Ҫ����ԣ���ˣ����������۽����ȱ��˵������
�����Щ�����ԣ�������������˵�һ��ԭ����������ѧ�о���������ͨ��ȱ������ѧ��ģ����Ͻ�����ṹ���������ѧ���㣬Ԥ�������ͼ�������ڽ�����ͼ��Ԥ���������������ѧ�����µĽ���ƽ����ṹ�����������ǿ�ȡ����Ͻ������Ԫ�صĽ���ƫ�ۼ���ЧӦ���Լ������ںϽ�Ԫ��������ԭ�ӿ��ܵ�����á�ͨ��������о�ʵ�������������Ⱥ��ʾ���Ͻ�Ԫ��Hf��Y��Ce����Pt��BC/TGO�����ǿ��ЧӦ�ͻ���[16-20]��������һ��ԭ����������ѧ�ɹ��ƹ㵽������������ϵ[21-23]�������������������������Ļ����ϣ���һ������������Ͻ�Ԫ��Zr��BC/TGO(��-Ni(Al)/��-Al2O3)�����ǿ��ЧӦ�����������������ͬ��Ԫ��Hf�Ľ���ЧӦ���жԱȣ�ΪBC�Ͻ���Ż�����ṩ��ѧָ��������������¼�����Ҫ�о����ݣ�
1) ����ȷ��1300 K�¦�-Ni(Al)/��-Al2O3������ͼ�ĵ��½��棻
2) ���ݻ�ȼ��㣬ȷ�����¶��µĽ���ƽ����ṹ��
3) ����ͱȽ�ƫ��ǰ�������빦(Wsep)�ı仯����������ƫ�۶Խ��ǿ�ȵ�Ӱ�죻
4) ����Zr��S����ƫ�۵��������Ϳ��ܵ�ƫ��·����
5) ��������������Zr��Ni����������S�Ķ���������
���м�����õ�һ��ԭ�������ܶȷ���ͨ�ó����VASP[24]�����н��������ܲ��ù����ݶȽ��ƣ��������֮�������ò���Vanderbilt�������ƣ�ƽ�沨չ�����ýϸ������ض�(400 eV)���й�Al��ȵļ��㣬���ð�������Alԭ�ӵ�2��2��2 ���Ħ�-Ni��������Ԩ������������6��6��6 Monkhorst-Pack (M-P) k-�����н��С��¶ȶ������Ĺ���ͨ��ֱ�ӳ����������Ӽ������г���ƹ���[25]��������������������-Ni(111)/��-Al2O3(0001)����ģ�ͣ�����8��Niԭ�ӡ�12��Alԭ�ӡ�6��Oԭ�ӡ�������12  ����ղ㣬����Ԩ���������ֲ���3��3��1 M-P k-�����������ð�������Zr-Sԭ�ӶԵ�3��3��3���� ��-Ni�ij������2��2��2 M-P k-�������м���Ϊ��ȫ��ԥ���㣬�����о�ΪHellmann-Feymannԭ�Ӿ���С��20 meV/(1eV/=1.6 nN)�������п�����诼���������������-���ӵ�������Լ�����ת�䡣
����ղ㣬����Ԩ���������ֲ���3��3��1 M-P k-�����������ð�������Zr-Sԭ�ӶԵ�3��3��3���� ��-Ni�ij������2��2��2 M-P k-�������м���Ϊ��ȫ��ԥ���㣬�����о�ΪHellmann-Feymannԭ�Ӿ���С��20 meV/(1eV/=1.6 nN)�������п�����诼���������������-���ӵ�������Լ�����ת�䡣
1 ����ṹ�����ǿ��
��������Ϳ����BC/TGO�����γɹ����У�BC�Ͻ��й��ܵ�AlԪ�����������������������Al2O3�㡣ͬʱ������AlԪ�صIJ������ģ�BC�Ͻ����ٽ����洦���ڲ���֯������Ӧת�䣬��ͨ��������(��/�»��/�á�)ת��ɵ���(��)����������������Al�������ĺ�Al2O3��IJ�������������������γɽӽ���ﵽ����ѧ�ȶ��Ħ�-Ni(Al)/��-Al2O3���档EVANS��[26]�Ե��Ͷ������Ϳ��ϵͳ�Ľ�����ϵ��ɼ�����Ԫ��Ļ��������������������ˣ���һ���һ��ԭ���о������������ķ���[14-15]��ͬ�����������о��������Ľ��ǿ�ȣ����ȿ������������̵�����ѧƽ�����������Ӽ������ƽ����ͼ��ʼ��
1.1 ����ƽ����ṹ��ȷ��
��������������ྦྷ�����IJ��죬�����γ�ʱ���ɱ������ʧ��λ��������һ���㹻������ͽ��泬����������ʧ��λ������Ȼ���������ڣ������¼���ļ��������ڦ�-Ni/��-Al2O3���渽�����������뾡����С�ı����������γɽӽ����빲��Ľ���ԭ�ӽṹģ�͡��ڼ������ǿ��(��������빦)ʱ�����ڽ���������������֮��������������������̶ȵ�����Ӧ��Խ�����빦��ֵ��Ӱ�졣
ͼ1��ʾΪ3�ֵ��ͽ���ԭ�ӽṹ���͵Ĺ�����ϵ(��ģ�͢�͢�)���ֱ����3�ֵ��͵Ľ���ȡ�����Ӧ������[16]�����У����Ц�-Ni(111)( ��)//��-Al2O3(0001)(1��1)ȡ���ϵ�Ľṹ��(ģ�͢�͢�)������Ľ���Niһ�����������Ӧ�䣬�����ƽ���(Nb/Al2O3)�ĸ߷ֱ�羵(HRTEM)�۲�[27]�Ϸ��ϡ��ݴ��жϣ�ģ�͢�͢��Ϊ����������ģ�͢����蹲��Ӧ���������С������ں��������в��á�
��)//��-Al2O3(0001)(1��1)ȡ���ϵ�Ľṹ��(ģ�͢�͢�)������Ľ���Niһ�����������Ӧ�䣬�����ƽ���(Nb/Al2O3)�ĸ߷ֱ�羵(HRTEM)�۲�[27]�Ϸ��ϡ��ݴ��жϣ�ģ�͢�͢��Ϊ����������ģ�͢����蹲��Ӧ���������С������ں��������в��á�
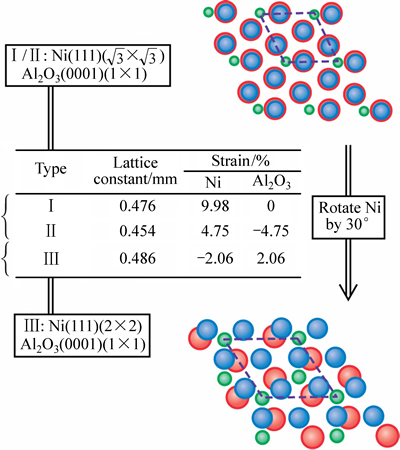
ͼ1 ���ͽ���ṹ�Ľ���ȡ�����Ӧ���ϵ(Ni��Al��O ԭ�ӷֱ��������̡���ɫ��ʾ)
Fig. 1 Typical commensuration types for ��-Ni/��-Al2O3 interface with different orientations and coherent strains (Ni, Al and O atoms are represented by dark blue, green and red spheres, respectively)
1.1.1 ����ԭ�ӻ�ѧ��ȵ�Ӱ��
BC/TGO�����ƽ����ṹ����ʵ������������(���¶Ⱥͻ�ѧ��Ӧ����)���������л�ѧ���տ�������ѹ��Ͻ��е�Al��ȱ�������ֱ�Ӿ�����ʵ�ʲ�����滯ѧ��Ӧ�ĸ�Ԫ��ԭ�ӵ���Ժ����������渽����ԭ�ӻ�ѧ�����ȡ��ο������ն�(Termination)�ĸ�����Լ���3�ֵ��͵Ľ���ԭ�ӻ�ѧ������ͣ������뻯ѧ������(Ni/Al2O3)����Al(Ni/AlAl2O3)��O��(Ni/OAl2O3)[16]���ֱ�����Ӧ�Ľ�����빦���������������ڽ���ȡ�����Ӧ�䣬����ԭ�ӻ�ѧ����Ǿ���������ǿ�ȵ���Ҫ����������ṹ���Ͳ�ͬ����ԭ�ӻ�ѧ���������ͬ��������빦��������50%��������ṹ������ͬ����ԭ�ӻ�ѧ��Ȳ�ͬ������ǿ�������ɴ��������ϡ���ģ��II����Ϊ������Al��O�ͽ���Ľ���ǿ�ȣ��ֱ������뻯ѧ�����Ƚ����4����6������Щ�������������BC/TGO�������������棬�κ����ӽ���ԭ�ӻ�ѧ��ȵļ����о������������������Dz��������ֱ�����Ӧ�Ľ����ܣ���ͨ�����������¶Ⱥ�Al���֮�������ѧ��ϵ���ɼ������ƽ����ͼ���Ӷ�����Ԥ���γɸ߽��ǿ�ȵĸ�Al��O�ͽ������������ѧ������
1.1.2 Al��������ƽ����ͼ�Ĺ�ϵ
����ȱ������ѧ�������Ƶ�������ѧƽ��������Ni/AlB2BOB3B�����ܵı���ʽ[28]
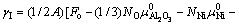
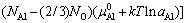 (1)
(1)
ʽ�У�Fo������������Helmholtz�����ܣ��ϱ� ��0����ʾ����Ԫ�Ĵ���̬��Ni�ͦ�i�ֱ��Ǹ���ԪԪ��i��ԭ�Ӹ����ͻ�ѧ�ƣ�A�ǽ������������ǰ����������֮����ڴ����������¶ȶԽ����ܵ�Ӱ����Ҫ���������һ���kT lnaAl������aAl�Ǧ�-Ni(Al)�����Al��ȣ��ɼ�������[25]
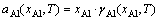 (2)
(2)
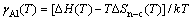 (3)
(3)
ʽ�У���Al�ǻ���Ni(Al)�Ͻ��е�Al������ӣ��ڽϵ�Ũ��(xAl��10%~15%)ʱһ�����Ϊ��Ũ���أ���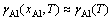 ��DH��DSn-c�ֱ�Ϊ����Alԭ�ӴӴ������ʹ��ܵ���-Ni(Al)���������������ʱ�ͷ����ر�[25]��ͼ2��ʾΪ����õ���Al����������¶ȵı仯��ϵ[29-32]�������ʵ��ֵ�Ƚϣ�����ֵ��ʵ��ֵ�ڸ��������ձ��ǺϽϺã����ڽϵ��¶�(980 K)ʱƫ����4�����������ҡ�����Ҫ�����ڽӽ�Al�۵���¶��£���������ѹ��ʵ��ֵ���̫�����֮�£����ۼ��������ѧԭ������������ʵ���������ޣ��ɺ��ǽϹ㷺���¶����䣬����Ԥ���Ϊ�ɿ������о��н���Ҫ��Ը�������Ϊ1300~1600 K��
��DH��DSn-c�ֱ�Ϊ����Alԭ�ӴӴ������ʹ��ܵ���-Ni(Al)���������������ʱ�ͷ����ر�[25]��ͼ2��ʾΪ����õ���Al����������¶ȵı仯��ϵ[29-32]�������ʵ��ֵ�Ƚϣ�����ֵ��ʵ��ֵ�ڸ��������ձ��ǺϽϺã����ڽϵ��¶�(980 K)ʱƫ����4�����������ҡ�����Ҫ�����ڽӽ�Al�۵���¶��£���������ѹ��ʵ��ֵ���̫�����֮�£����ۼ��������ѧԭ������������ʵ���������ޣ��ɺ��ǽϹ㷺���¶����䣬����Ԥ���Ϊ�ɿ������о��н���Ҫ��Ը�������Ϊ1300~1600 K��
��Բ�ͬ�Ľ�����ṹ��������������¶��½�������Al��ȵĶ�Ӧ��ϵ���Ϳ��Եõ�����ƽ����ͼ��һ�����½��档ͼ3��ʾΪͨ��ʽ(1)~(2)����õ���T=1300 K���½��漰��Ӧ�Ľ�����ṹ�������������γɺ��渽��Al�Ĵ���Ũ�ȷ�ΧΪ1�� xAl��15%�����Al������ӵļ������������ƶϽ��渽��Ni(Al)һ��AlԪ�صĴ��»�ȷ�Χ�����ݵ��½�����ͼ�������жϸ��¶��½���ƽ����Ӧ�Ǹ�Al��(Ni/AlAl2O3)�����������뻯ѧ��������硣
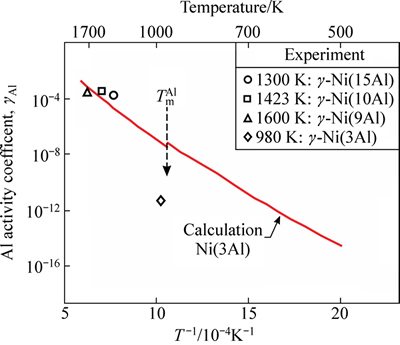
ͼ2 ����Ԥ���Al����������¶ȵı仯��������Ӧʵ��ֵ[29-32]�ıȽ�
Fig. 2 Comparison of calculated Al activity coefficient in ��-Ni(Al) and experimental values[29-32]
��ͼ3���ɿ��������㹻�ߵ�Al���(����������)�����£�����������ֱ���������������仯����(Ni3Al)��������Al��Ȳ��Ͻ���(��Ӧ������ѹ�IJ�������)�����¶��½���ƽ���������ת��Ϊ���뻯ѧ��������(Ni/Al2O3)����O��(Ni/OAl2O3)������������(NiAl2O4)��ֱ��������(NiO)�����и������������������������γɣ���һЩ���ⷽ���Ʊ���BC/TGO�����϶��õ���ʵ��֤ʵ[30, 33]��
���ڻ����е�����ѹ��Ͻ��е�Al���֮�����һ���Ļ����ϵ������������������Ӧ��Al(solid)+O2(gas)=Al2O3(solid)������ѧƽ�������Ƶ�[24]����ˣ����ݽ���ƽ����ͼ�����Խ�һ��Ԥ���γɲ�ͬ������ṹ����Ķ�Ӧ����ѧ��������������ʵ����Ա�����ͽ����ڶ�ʵ������ö�������Լ�ָ��ʵ���Ʊ����յ�����Ż����ڴ˲�������
1.2 ����ƫ�������ǿ��
�ڼ����Ͻ�Ԫ��Zr�Ľ���ƫ��ʱ��Ϊ�����������ͬʱ�������ֽ���(����Al�ͺ����뻯ѧ�������ͽ���)��ƫ����Ϊ���������Ƚ���Խ�����빦��Ӱ�졣
1.2.1 ����ƫ���ܺ�ƫ��·���Ĺ�ϵ
��ǰ������ͬ���Ļ��壬�ڲ�ͬ��������(�¶Ⱥ�����)�µõ��Ľ���ƽ������ܲ�ͬ����ˣ��Ͻ������Ԫ�صĽ���ƫ������Ҳ����Ӧ��ͬ���Ӷ�ֱ��Ӱ�쵽���ǶԽ����Ӱ������á�����ƫ������������ͨ������ͱȽ�ƫ��ǰ����泬�����ܵı仯(������ƫ����)����������ֵ�Ľ���ƫ���ܴ����ӻ���������ƫ�����ƣ���֮��Ȼ���Ƚ�ͬһԪ�ضԽ��治ͬλ�õ�ƫ���ܣ�����ȷ����Ԫ�ص�ƫ��·�����Ƚϲ�ͬԪ�صĽ���ƫ���ܣ�����ȷ��Ԫ��֮���ƫ������Ȩ��
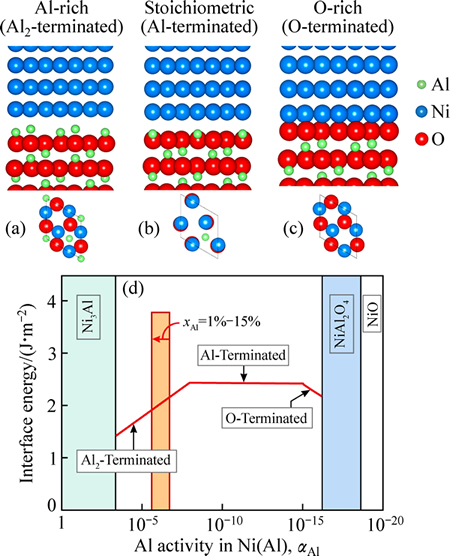
ͼ3 ��Al��(Ni/AlAl2O3)�����뻯ѧ�����(Ni/Al2O3)�Լ���O��(Ni/OAl2O3)�Ľ���ԭ�ӽṹ�Լ�����õ��Ľ���ƽ����ͼ��T=1300 K�ĵ��½���(ͼ�г�ɫ�����������֪�Ľ��渽��Al��ȵĴ��·�Χ)
Fig. 3 Atomic structures of Al-rich (Ni/AlAl2O3) (a), stoichiometric (Ni/Al2O3) (b) and O-rich (Ni/OAl2O3) interfaces (c) and calculated interfacial phase diagram for T=1300 K (d) (Approximate variation range of near-interface Al activity is represented as orange bar)
ͼ4��ʾΪ����Ԥ���Ԫ��Zr�Խ����ƫ������������֮ǰ����Sƫ���ļ�����[12, 14]�Աȡ������ʾ��1) �������뻯ѧ�����Ƚ��棬����S��ǿ�ҵ�ƫ���������ڽ������϶λ(SI)��Ni�û�λ(SNi)��S��ƫ���ֱܷ�Ϊ1.74��1.44 eV/atom��������Ԫ��Zr�Ľ���ƫ��������ǿ���ڽ�����϶λ(ZrI)��Ni�û�λ(ZrNi)��Zr��ƫ���ֱܷ�ﵽ2.34��2.02 eV/atom��2) ���ڸ�Al�ͽ��棬S��ƫ��������ͣ��ڽ���Ni�û�λ(SNi)����϶λ(SI)�ϣ�S��ƫ���ܽ��ֱ�Ϊ0.44 ��0.20 eV/ atom����Զ��ԣ�Zr��Ȼ���к�ǿ��ƫ����������Al�û�λ(ZrAl)��ƫ����Ϊ1.08 eV/atom���Ա�֮ǰ�ļ�����[17]�����Է��֣�������������ϣ�Zr��ƫ��������ͬ���Hf��ǿ���Ҿ�����ǿ������S��S�ܷ��ڽ���ƫ���������ò�ȡ�������ڵִ�����Zrԭ��ռλ�������뻯ѧ�����Ƚ���(��ͼ4(b))Ϊ����ƫ�������������S�����Ⱥ�ռ��Ni�û�λ(SNi)����϶λ(SI)����ϵ���ܽ�һ������0.87 ev/atom�������������ƫ��Zr���ڣ�����S��ƫ����Ȼ���Լ������У�����ռNi�û�λ(SNi)���ڽ����ϳ���Zr��S�ġ���ͬƫ��������ϵ���ܽ�һ�����͡���ͼ4(a)���Կ��������ƵĹ�ͬƫ��Ҳ�����ڸ�Al��Ľ����ϳ��֡�����ƫ��(������ͬƫ��)�Խ���ǿ�ȵ�Ӱ�죬����ͨ��ֱ�Ӽ�����Ӧ�Ľ�����빦����������
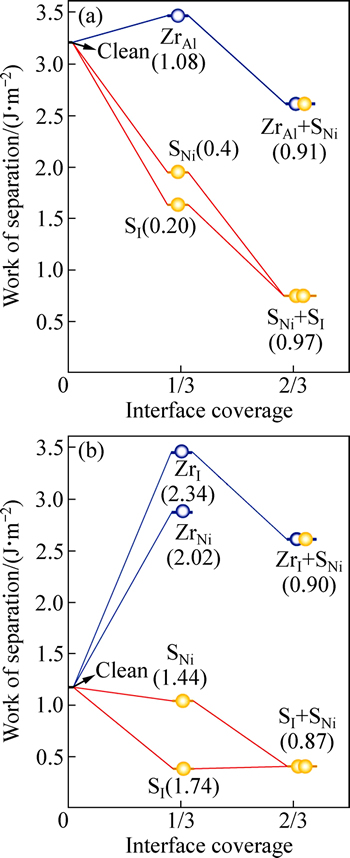
ͼ4 ��Al������뻯ѧ��������Ľ�����빦(Wsep)�����ƫ���ȵı仯(����ƫ������Ni(111)����ÿNiԭ��Ϊ��λ�������е����ִ�����ƫ�����̶�Ӧ�Ľ���ƫ����(��λ��eV/atom))
Fig. 4 Work of separation, Wsep as function of interfacial coverage for Al-rich (a) and stoichiometric interfaces (b) (Interfacial coverage is valued as per Ni atom on Ni(111) layer, values in parentheses correspond to associated heats of segregation for each segregation step (Unit: eV/atom))
1.2.2 Ԫ��ƫ���Խ���ǿ�ȵ�Ӱ��
�Ƚ�ƫ��ǰ�����ṹ�ı仯��������Ӧ�Ľ�����빦(Wsep)�����Զ�������Ԫ��ƫ���Խ�����ǿ�ȵ�Ӱ�졣�йؽ�����빦�ļ������ɼ�ͼ4��5�����ڸ���Ȥ�Ľ��棬���Խ����Ӧ�ļ۵����ܶȷֲ���ͼ(��ͼ6)�����Է������渽�����ӳɼ����ص㣬�Լ��Ͻ������Ԫ��ƫ���Խ��滯ѧ�ɼ���Ӱ�졣
��ͼԤ��Ϊ����ѧƽ����ĸ�Al�࣬�䴿��������нϸߵĽ�����ǿ��(Wsep=3.20 J/m2)��Լ3�������뻯ѧ�����Ƚ����ǿ��(1.14 J/m2)���������ڸ�Al��������γ��к�ǿNi��Al������(��ͼ6(a))��Զǿ�����뻯ѧ������������ϵ�Ni��O��(����-���ӻ�ͼ�����ͼ6(b))��������������ϣ��ر��������뻯ѧ������������ϣ�������Խ���ǿ��Σ�����ɽ��ͽ�����빦��60%~70%(��ͼ4��5)��������Ľ���ƫ����������ԭ�������ǿ��Ni��O���������Ĺ��ۼ�S��O�����������Ӽ�S��Al���滻(��ͼ6(d))�����½���������ֵ��ע����ǣ��Ͻ�Ԫ��Zr�Ľ���ƫ��ЧӦ��ͬ���HfԪ��[17]�dz����ơ�Zrƫ�۶Խ����ǿ�����ã�����Խ��������뻯ѧ�����Ƚ����ϱ��ֵ���Ϊ����(��ͼ4(b)��5)���ر��ǵ�Zrƫ������϶λ��(ZrI)����ʹWsep����Լ300%����ϸ�۲����õ��Ļ�̬����ԭ�ӽṹ��֪������������ǿ��Ч����ʵ����Դ���ڽ���ԭ�ӽṹ�ĸı䣺Zr�����뻯ѧ�����Ƚ������϶λ��ƫ��(ZrI)��ǡ�ɸ���Zr�ڸ�Al������ϵ�Al�û�λƫ��(ZrAl)�Ľ���ṹ����Ӧ�Ľ���۵����ܶ���ͼ(��ͼ6(c)) �����ʾ��Zr�ܹ�ͬʱ����������Ni��O�γ��¼����¼�����Զǿ��ͼ6(b)�е�Ni��O�����ӽ�ͼ6(a)�е�Ni��Al���������Ӷ�ʹԭ�����������뻯ѧ�����Ƚ���ǿ��������3�����ﵽ��Al���������۽��ǿ�ȡ���Ȼ��Zr�ܹ�ֱ�Ӳ������ɼ����Ӷ�ǿ�����档ֵ��ע����ǣ�ͼ4(b)�������ʾ��Zr�Խ����ǿ�����ã����������ƫ����S�ƻ�����������ˣ�������������棬Zr��S�Ĺ�ͬƫ����Ҫ�����ܵر��⡣ֻ���跨��S��Ч�ض����ڻ����У��������ڽ����ƫ�������п�������ȵط���Zr��ǿ�����á�
2 �����ڵ����ʶ���
������ֱ�ӱ�������Ͻ���ԭ�Ӽ������ã�Ŀǰ��ʵ���⼼���ֶ�������ʵ�֡����õ�һ��ԭ��ֱ�Ӽ���ͱȽ�Zr-Sԭ���ڦ�-Ni(FCC)�����е������(2.5 )���������������Զ�����ϵ�ϵͳ���ܣ������ֵ�ɶ�����ӳZr-S֮������õ�ǿ��������֮��Ϊ����Zr-Sԭ�Ӷ��ڦ�-Ni�����о�������ѣ���֮��Zr-Sԭ�Ӷ��ڦ�-Ni�����о������ȶ�������S�ܹ���Zr��Ч��������Ȼ�����ܲ�ֵԽ��������������Խǿ��ʵ�ʼ����У�ѡ����FCC�����еĵ�����ھ�����Ϊ����Զ����(ԼΪ8.5 )������ȷ����ʱ����ԭ�Ӽ���㹻Զ������ÿɺ��Բ��ơ�
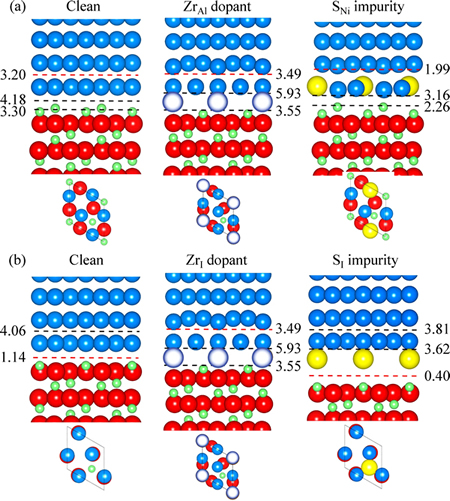
ͼ5 Ԫ��ƫ���Ը�Al���������뻯ѧ�����Ƚ���Ľ��ǿ��
Fig. 5 Segregation structures and Wsep for Al-rich (a) and stoichiometric interface (b) at 1/3 monolayer coverage of Zr and S (Ni, Al, and O atoms are represented in dark blue, green and red, respectively. Zr and S atoms are represented in bright blue and yellow, respectively)
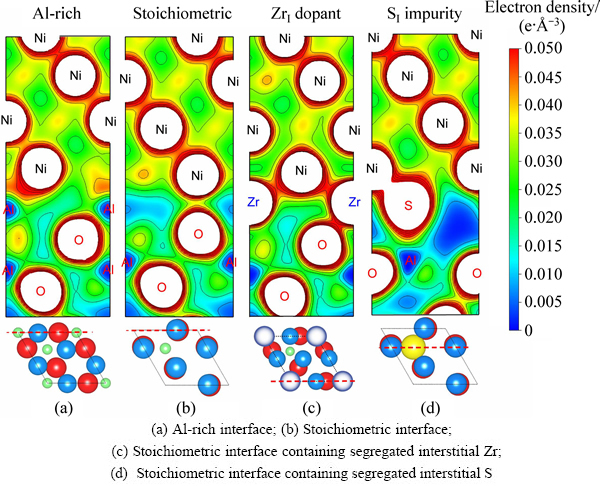
ͼ6 ����۵����ܶ���ͼ
Fig. 6 Interfacial valence electron density contours (Dash-dot lines at bottom correspond to positions of contour planes in top-views, (Unit: e/3))
��ؼ������ɼ�ͼ7(a)�������г�ֿ����¶ȶԾ�������صĹ���(�������ӣ�Phonons)�Լ��ȵ���ЧӦ[34]����ͼ7(a)��������0K�£����������κ��¶�ЧӦʱ��Ni�����е�Zr��S���γ��ȶ���ԭ�Ӷ�������ý�ǿ������֮��ԼΪ-0.21 eV/atom�������¶ȵ����ߣ�����������ЧӦ�ᵼ��ԭ�Ӽ���������¾������������ȵ���ЧӦ�������ع�ͬ���ã����һ��Ӱ��Zr-S֮�������á�����������ڸ���Ȥ��������������(1300~1600 K)��Zr���ܽ�ǿ�ض���S��������Ϊ-0.2~0.44 eV/atom����ͬ���Hf���������ӽ�[17]��ͼ7(b)��ʾΪ������Ni������Zr-Sԭ�ӶԵļ۵��Ӿ������ֲ�(Electron localization function, ELF[35])����Ni-Zr�����ľ��ȵ����ܶȱ��������£������ܶ���Ҫ��S��������Zr������Լ��У���ʾZr-S���������Ҫ�����Ӽ����ʣ������ܻ�������Ĺ��۳ɷ֡�����̬�ܶȷ���Ҳ֤ʵ������Zr(5d,6s)-S(3p)����ӻ�����ͬ��Hf��ͬ��Zr��Sԭ��֮����ڼ۵��Ǩ�ƣ������ڦ�-Ni�������ܹ���Ч��������S���ۻ��ơ�
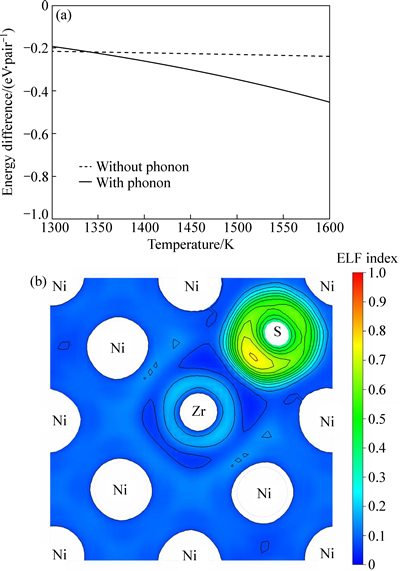
ͼ7 ��-Ni������Zr-Sԭ�Ӷ���������¶������¶ȵı仯��Zr-Sԭ�ӶԸ����ĵ��Ӿ���������ͼ
Fig. 7 Change of interaction energies of Zr-S pair in ��-Ni with temperature at high temperatures (a) and electron localization function (ELF) contours around Zr-S pair (b) (Dashed lines refer to interaction energies calculated at 0 K, reflecting low-temperature pair attraction)
3 ����
1) ����Ϳ����BC/TGO�ؼ�����Ľ���ṹ����������(Al��Ȼ�O��ѹ)�ͻ����¶Ⱦ��������ݼ���õ��Ľ���ƽ����ͼ���ڸ���Ȥ���¶�����(1300~1600 K)������ƽ����Ϊ��Al�࣬���������뻯ѧ�����������硣��Al�������ǿ�Ƚϸ�(Wsep=3.20 J/m2)��Լ���������뻯ѧ�����Ƚ���(1.14 J/m2)��������Sƫ���������ֽ����ϣ����ɽ��ͽ�����ǿ�ȴ�60%~70%��
2) �Կ��ܹ�������ֽ����࣬�Ͻ�Ԫ��Zr������ǿ�ҵ�ƫ����������ƫ��������ͬ��Ԫ��Hf��ǿ��
3) Zr�Ͻ����������ڽ���ǿ������ǿ��������ͬ��Ԫ��Hf���ƣ���Ҫ����3�֣���Ni��������Ч��������S����S�Ľ���ƫ�����ڽ��洦�û�ƫ������S����SЧӦ�Խ����ֱ��Σ������ֱ�Ӳ������ɼ���ǿ�������ϡ�
4) ���ڽ�����������뻯ѧ�����Ƚ��棬Zr�Ͻ�����������빦Լ3��(Wsep=3.5 J/m2)��������Al�ͽ�������۽��ǿ��(3.20 J/m2)��
REFERENCES
[1] EVANS A G, MUMM D R, HUTCHINSON J W, MEIER G H, PETTIT F S. Mechanisms controlling the durability of thermal barrier coatings[J]. Progress in Materials Science, 2001, 46(5): 505-553.
[2] GLEESON B. Thermal barrier coatings for aeroengine applications[J]. Journal of Propulsion and Power, 2006, 22(2): 375-383.
[3] ������, �� ��, ������. ����Ϳ����о���״�뷢չ����[J]. �й���ɫ����ѧ��, 2007, 17(1): 1-13.
LIU Chun-bo, LIN Feng, JIANG Xian-liang. Current state and future development of thermal barrier coating[J]. The Chinese Journal of NonferrousMetals, 2007, 17(1): 1-13.
[4] SMIALEK J L, JAYNE D T, SCHAEFFER J C, MURPHY W H. Effects of hydrogen annealing, sulfur segregation and diffusion on the cyclic oxidation resistance of superalloys: A review[J]. Thin Solid Films, 1994, 253(1/2): 285-292.
[5] GAUDETTE F G, SURESH S, EVANS A G. Effects of sulfur on the fatigue and fracture resistance of interfaces between gamma-Ni(Cr) and alpha-Al2O3[J]. Metall Trans A, 2000, 31(8): 1977-1983.
[6] KIELY J D, YEH T, BONNELL D A. Evidence for the segregation of sulfur to Ni-alumina interfaces[J]. Surf Sci, 1997, 393(1/3): L126-130.
[7] FUNKENBUSCH A W, SMEGGIL J G, BORNSTEIN N S. Reactive element-sulfur interaction and oxide scale adherence[J]. Metall Trans A, 1985, 16(6): 1164-1166.
[8] KHANNA A S, WASSERFUHR C, QUADAKKERS W J, NICKEL H. Addition of ytterium, cerium, and hafnium to combat the deleterious effect of sulfur impurity during oxidation of an Ni-Cr-Al alloy[J]. Mat Sci Eng, 1989, A120(1):185-191.
[9] HOU P Y. Segregation phenomena at thermally grown Al2O3/Alloy interfaces[J]. Annual Review of Materials Research, 2008, 38: 275-298.
[10] BARRETT C A. Effect of 0.1 at.% zirconium on the cyclic oxidation resistance of ��-NiAl[J]. Oxidation of Metals, 1988, 30(5/6): 361-390.
[11] HAMADI S, BACOS M P, POULAIN M, SEYEUX A, MAURICE V, MARCUS P. Oxidation resistance of a Zr-doped NiAl coating thermochemically deposited on a nickel-based superalloy[J]. Surface and Coatings Technology, 2009, 204(6/7): 756-760.
[12] OZFIDAN I, CHEN K Y, FU M, Effects of Additives and Impurity on the adhesive behavior of the NiAl(110)/Al2O3 (0001) interface: An ab initio study[J]. Metallurgical and Materials Transactions A, 2011, 42(13): 4126-4136.
[13] ��־��, ��Զ��. ճ�Ӳ���Cr������Ϳ����������ӵ�Ӱ��[J]. �й���ɫ����ѧ��, 2004, 14(9): 1477-1482.
LI Zhi-lin, WU Yuan-qi. Influence of bonding layer Cr content of thermal barrier coatings on its interface conjunction factors[J]. The ChineseJournalof NonferrousMetals, 2004, 14(9): 1477-1482.
[14] CARLING K M, CARTER E A. Effects of segregating elements on the adhesive strength and structure of the ��-Al2O3/��-NiAl interface[J]. Acta Materialia, 2007, 55: 2791-2830.
[15] JARVIS E A, CHRISTENSEN A, CARTER E A. Weak bonding of alumina coatings on Ni (111)[J]. Surface Science, 2001, 487(1/3): 55-76.
[16] SMITH J R, JIANG Y, EVANS A G. Adhesion of the gamma-Ni(Al)/alpha-Al2O3 interface[J]. International Journal of Materials Research (formerly Z. Metallkd.), 2007, 98(12): 1214-1221.
[17] JIANG Y, SMITH J R, EVANS A G. First principles assessment of metal/oxide interface adhesion[J]. Applied Physics Letters, 2008, 92(14): 141918.
[18] JIANG Y, SMITH J R. Pt effects in gamma-Ni(Al)/alpha-Al2O3 adhesion[J]. Journal of Materials Science, 2009, 44(7): 1734-1740.
[19] �� ��. ���ڵ�һ��ԭ����ʾ����Ԫ����������Ϳ��ؼ������ǿ������[J]. �й���ɫ����ѧ��, 2011, 21(6): 1463-1472.
JIANG Yong. Strengthening mechanisms of reactive-element Hf on the key interface in thermal barrier coating systems from the first-principles[J]. The ChineseJournalof NonferrousMetals, 2011, 21(6): 1463-1472.
[20] LAN G Q, WANG Y R, JIANG Y. Effects of rare-earth dopants on the thermally grown Al2O3/Ni(Al) interface: the first-principles prediction[J]. Journal of Materials Science, 2014, 49: 2640-2646.
[21] LAN G Q, JIANG Y, YI D Q, LIU S J. Theoretical prediction on microstructure evolution during the internal oxidation fabrication of metal-oxide composites: the case of Cu-Al2O3[J]. RSC Advances, 2013, 3(36): 16136-16143.
[22] LAN G Q, JIANG Y, YI D Q, LIU S J. Theoretical prediction of impurity effects on the internally oxidized metal/oxide interface: The case study of S on Cu/Al2O3[J]. Phys Chem Chem Phys, 2012, 14(31): 11178-11184.
[23] �� ��, ����ǿ, ������, ������. ������Cu/Al2O3��������ѧ������ЧӦ[J]. �й���ɫ����ѧ��, 2013, 23(11): 3154-3164.
JIANG Yong, LAN Guo-qiang, WANG Yi-ren, ZHOU Song-song. Interfacial thermodynamics and impurity effects on internally oxidized Cu/Al2O3 interface[J]. The ChineseJournalof NonferrousMetals, 2013, 23(11): 3154-3164.
[24] KRESSE G, FURTHM LLER J. VASP manual[EB/OL]// http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html
LLER J. VASP manual[EB/OL]// http://cms.mpi.univie.ac.at/vasp/vasp/vasp.html
[25] JIANG Y, SMITH J R, EVANS A G. Temperature dependence of the activity of Al in dilute Ni(Al) solid solution[J]. Physical Review B, 2006, 74(22): 224110.
[26] EVANS A G, CLARKE D R, LEVI C G. The influence of oxides on the performance of advanced gas turbines[J]. J Euro Ceram Soc, 2008, 28(7): 1405-1419.
[27] VITEK V, GUTEKUNST G, MAYER J, RUHLE M. Atomic-structure of misfit dislocations in metal-ceramic interfaces[J]. Philosophical Magazine A, 1995, 71(6): 1219-1239.
[28] ZHANG W, SMITH J R, EVANS A G. The connection between ab initio calculations and interface adhesion measurements on metal/oxide systems: Ni/Al2O3 and Cu/Al2O3[J]. Acta Materialia, 2002, 50(15): 3803-3816.
[29] ESKOV V M, SAMOKHVAL V V, VECHER A A. Thermodynamic properties of Al-Ni hard alloys[J]. Russ Metall, 1974, 2: 118-119.
[30] STEINER A, KOMAREK K L. Thermodynamic activities of solid nickel-aluminum alloys[J]. Trans Metall Soc AIME, 1964, 230(4): 786-788.
[31] OFORKA N C. Thermodynamics of nickel-aluminum alloys[J]. Indian J Chem A, 1986, 25(11): 1027-1029.
[32] HILPERT K, MILLER M, GERADS H, NICKEL H K. Thermodynamic study of the liquid and solid alloys of the nickel-rich part of the Al-Ni phase-diagram including the AlNi3 phase[J]. Ber Bunsenges, Phys Chem, 1990, 94(1): 40-47.
[33] DAROONPARVAR M, YAJID A M M, YUSOF N M, FARAHANY S, HUSSAIN M S, BAKHSHESHI -RAD H R, VALEFI Z, ABDOLAHI A. Improvement of thermally grown oxide layer in thermal barrier coating systems with nano alumina as third layer[J]. Transactions of Nonferrous Metals Society of China, 2013, 23(5): 1322-1333.
[34] JIANG Y, LIU R. Gettering of S in Ni from first-principles[J]. Scripta Materialia, 2010, 62(10): 782-785.
[35] BECKER A D, EDGECOMBE K E N. A simple measure of electron localization in atomic and molecular systems[J]. Journal of Chemical Physics, 1990, 92(9): 5397-5403.
Strengthening effects and mechanisms of micro-alloying Zr on key interface in thermal barrier coating systems
LAN Guo-qiang1, 2, JIANG Yong1, 2, 3, 4, JIANG Liang3, 4
(1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Key Laboratory of Nonferrous Materials, Ministry of Education, Central South University, Changsha 410083, China;
3. National Key Laboratory for Powder Metallurgy, Central South University, Changsha 410083, China;
4. Shenzhen Research Institute, Central South University, Shenzhen 518057, China)
Abstract: Bond-coat/thermally-growth-oxide interface is the key interface in high-temperature thermal barrier coating systems. The first-principle interface thermodynamics modeling combined with density functional theory calculations were performed to investigate the effects of micro-alloying Zr on the ��-Ni(Al)/��-Al2O3 interface structure, segregation, and adhesion, and pinning impurity (S) in the matrix. The results show that Zr plays very similar roles as Hf. The addition of Zr substantially improves the adhesion through three strengthening mechanisms: pining S in bulk Ni(Al), displacing S from its interstitial interface sites and directly enhancing the interfacial binding. The binding strength of the weak stoichiometric interface can be enhanced by up to a factor of 3.
Key words: thermal barrier coating; interface adhesion; micro-alloying; zirconium; first-principles
Foundation item: Project(51171211) supported by the National Natural Science Foundation of China; Project (2012AA03A514) supported by the National High Technology Research and Development Program of China; Project(JCYJ20140509142357196) supported by the Shenzhen Science and Technology Program, China
Received date: 2015-03-03; Accepted date: 2016-07-04
Corresponding author: JIANG Yong; Tel: +86-731-88836320; E-mail: yjiang@csu.edu.cn
(�༭ ����)
������Ŀ��������Ȼ��ѧ����������Ŀ(51171211)�����Ҹ����о������ƻ�������Ŀ(2012AA03A514)�������пƼ��ƻ���Ŀ(JCYJ20140509142357196)
�ո����ڣ�2015-03-03�������ڣ�2016-07-04
ͨ�����ߣ��� �£����ڣ���ʿ���绰��0731-88836320��E-mail��yjiang@csu.edu.cn
ժ Ҫ����Ը�������Ϳ���йؼ��Ĺ��ɲ�/����������棬ͨ����һ��ԭ����������ѧ��ģ���ܶȷ������㣬�������Ԫ�ض�g-Ni(Al)/��-Al2O3����ṹ��ƫ�����ͽ����ϵ�Ӱ�죬�Լ��ڻ����ж�����(��)�Ķ����������ʾ��ﯾ�����ͬ��Ԫ�������ƵĽ���ǿ�����ã���ǿ���������Թ���Ϊ3�֣����ڻ�������Ч����������������к�����ƫ�����ڽ��洦�û����Լ���ƫ����Խ����Σ���Լ��ڽ���ƫ����ֱ��ǿ�������ϣ���ʹ���������뻯ѧ�����Ƚ���ķ��빦���3����
