
��п��ṹAlSb��Ƕ��﮻���
�� ��, ����, �ռ���, ¬����
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ, ��ɳ 410083)
ժ Ҫ��
���õ�һԭ������ƽ�沨���㷽���о�������п��ṹAlSb��Ƕ��﮻����� ͨ�����㲻ͬǶ���Lix(AlSb)(0��x��2)��Li2+yAl1-ySb(0��y��1)���γ���(��E), �������Ӧ������ѧԭ����ɵó���LiǶ��AlSbʱ�ĵ�ѹ������������ͼ, �ɴ˽�һ��ȷ�������Ƕ��AlSb�Ľṹʱ, ������ռ��AlSb�еļ�϶λ��, Ȼ������LiǶ����������, ������ȡ��AlSb�е�Alλ�ôӶ��γ�Li3Sb�ࡣ ͨ������LiǶ��AlSbǰ����ܴ��ṹ��̬�ܶ�ͼ���Է���, AlSb�ĵ�������������LiǶ���������Ӷ�����, ��Liռ��AlSbȫ����϶λ�ú�ﵽ��ֵ, ��Li��һ�����Alʱ����������֮���͡�
�ؼ���: AlSb; ��п��ṹ; ��һԭ��; ����ƽ�沨; ����ӵ�� ��ͼ�����: TG146.2
���ױ�ʶ��: A
Lithiation/delithiation mechanism of AlSb with zinc-blende structure
YAN Jian, SU Yu-chang, SU Ji-tao, LU Pu-tao
(School of Materials Science and Engineering,Central South University, Changsha 410083, China)
Abstract: The lithiation/delithiation mechanism of AlSb with zinc-blende structure was investigated by the first-principles pseudopotential plane wave method. The plot of potential vs specific capacity of Li intercalated into AlSb can be drawn by calculating the formation energies(��E) of different types of lithiated phases of Lix(AlSb)(0��x��2) or Li2+yAl1-ySb(0��y��1) in combination with the corresponding thermodynamic principles. It is found that Li first occupies the interstitial sites when intercalated into AlSb, and followed by substituting for Al sites with the increase of Li intercalation capacity to form the phase of Li3Sb. Through analyzing the band structure and the density of states of AlSb before and after lithiation, it is shown that the conductibility of AlSb increases with the increase of Li intercalation capacity, and reaches the peak value when Li occupies all the interstitial sites of AlSb, followed by decreasing when Li begins to substitute for Al.
Key words: AlSb; zinc-blende structure; first-principles; pseudopotential plane wave; lithium-ion batteries
����ӵ����Ϊһ��ȫ�µ���ɫ��ѧ�����, ���б������ߡ� ��ص�ѹ�ߡ� �����¶ȷ�Χ���� �������������ŵ�, ����ܵ����ڶ��о��ߺ��̼ҵ�����[1]�� Ŀǰ����������ҵ������������ӵ�ظ���������Ҫ�����ڸ������͵�̼����, ����Ȼʯī�� CN�Ƚ�̼�� AN�Ƚ�̼�ȡ� ����������Щ̼���ϴ����ű������͡� �״γ�ŵ�Ч�ʵ͡� �л��ܼ���Ƕ��Ȳ���[2], �������������ҪѰ��һ�����ܸ��õķ�̼�������ϡ� ��1997��Idota��[3]������һ�����͵ľ��и߱�����������ѭ�����ܵķǾ��������������︺������(TCO)��, ��������������һ���ܵ�����о��ߵĹ�ע�� ��������Sn����Li�γɺ�����dz��ߵ�Li22Sn5�����仯����, ��ƽ��1mol��Sn������4.4mol��Li, ����Ӧ����������������Ϊ790mA��h/g, ��ʯī���۱�����(372mA��h/g)��2���ࡣ ��Al-Li��Ԫ��ͼ[4]��֪, Al��Li�����γ�3���ȶ��Ľ����仯����: LiAl�� Li3Al2��Li9Al4�� ���, ��Ҳ����ϣ����Ϊ����ӵ�ظ������ϵ�, ������Ƕ�����Ҳ�DZȽϸߵ�, ��߿��Դﵽƽ��1mol��Al����2.25mol��Li�� ��Ȼ��û���������ߵ�Ƕ�����, ���ǿ��ǵ��������ԭ������(26.98)ԶԶС���������ԭ������(118.69), ��������������������������(2234mA��h/g)��ԶԶ�����������ġ� ��ʹ���Ƕ���������γ�LiAl��ʱ, ������������������Ҳ�ߴ�993mA��h/g, �������������������������������, ����, ����������һ�ֺ��з�չǰ���ĸ������ϡ�
Hamon��[5]�����������������Ʊ���0.1��m��1��m�������Ƭ, ͨ���绯ѧ����������Щ����Ƭ�������������ߴ�1000mA��h/g����, ��������Ƕ�����������ľ��������, ʹ�����ǵ�ѭ�����ܻ�Զδ�ﵽʵ�û���Ҫ�� ���Hamon�Ƚ�������������������ڶ������Ԫ�����γɽ����仯����������, ���ɴ�����������������ϵ�ѭ������, ���ѵõ�Jeong��[6]��Lindsay��[7]��֤ʵ�� ʵ����, �ڴ���������������ڶ������Ԫ�����γɽ����仯�����������ܹ���������������ӵ�ص�ѭ�����ܲ���������������������, �������Ľ�������������Ҳ�����õ�, ��NiSb2��ѭ�����ܾͱȴ�Sb��ѭ�����ܺ�[8]��
��һԭ�����㷽����ȴ���ʵ���нϴ������, ������Ҫ���д������ظ���ʵ��, ֻ��Ҫ֪����ɶ�����ϵͳ��ԭ�������;���ṹ����, �Ϳ���Ԥ֪��ϵͳ�Ķ�����������, ��ƽ��������� ����ܡ� �������ܼ��������ȶ��Եȡ� Ceder�����������[9-11]�����õ�һԭ��ϵͳ���о�������ӵ������������-﮹���������������Ƕ�������, �ó�����ʵ�����dz��Ǻϵ�����Ԥ�⡣ ����ĿǰΪ��, �йظ������ϵ��ⷽ����о�ȴ�Ƚ��ټ��� Ϊ�˱�����������õ�һԭ������ƽ�沨�����о�����ӵ���þ�����п��ṹ��AlSb�������ϵ�Ƕ��﮻�����
1 ��һԭ�����������۱���
��һԭ����ָ�ڲ���-�±���Ĭ(Born-Oppenheimer)���ƺ�����-����(Hartree-Fock)���ƵĻ�����, �ڼ����н���ʹ�����ʿ˳���h�� ��������m�ͻ������e��3��������������, �Լ�ԭ�ӵĺ�������Ų�, ���������ɵ��ڵľ������, ͨ����Ǣ��������������ϵͳ��Ѧ���ɷ����Եõ�ϵͳ�Ļ�̬���ܡ�

���ڵ������ڷ�����(Fermions), ��������������ԭ��(Pauli exclusion principle)�ͷ���-������ͳ��(Fermi-Dirac Statistics), ����ʽ(1)�еĽ��Ƴ̶ȹ���, �����˵���֮��Ľ���-���ЧӦ, ʹ�ü���ľ����ܵ���һ�������ơ� Ϊ�˽��������, Hohenberg��Kohn��1964��������������ܶȷ�������[12](Density Functional Theory, DFT)�� ������뷨����Ϊԭ�ӡ� ���Ӻ���Ļ�̬�������ʿ����õ����ܶȺ���n(r)������, Ҳ���ǿ��Թ��Ϊ�������������������
����һ: ����������ȫͬ������ϵͳ�Ļ�̬�����ǵ����ܶȺ���n(r)��Ψһ������
������: ��������E[n(r)]�ڵ���������������¶���ȷ�ĵ����ܶȺ���n(r)ȡ��Сֵ, ������ϵͳ�Ļ�̬������
�ɴ˿ɼ�, DFT���������˽�����������Ϊ��������������ۻ���, ͬʱҲ��Ϊ������Ӻ���ĵ��ӽṹ�����ܵ��������ߡ� ��DFT�����, Kohn��Sham(������)[13]�㽫�������۾��廯��, �ó���Kohn-Sham���ܷ�������

������ʽ����
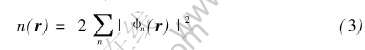
ʽ�� ����2��Դ�ڵ��ӵ�������������
ֻҪ֪�����Ӽ�������-�����ܷ���EXC[n(r)]�ľ�����ʽ, ��ôʽ(2)�Ϳ���ͨ����Ǣ�����Եõ�������ϵͳ�Ļ�̬���ܡ� Kohn��Shamͨ�������ܶȽ���(Local Density Approximation, LDA)�õ���EXC[n(r)]��һ�����Ʊ���ʽ�� ��LDA��, �����ݶȽ���(Generalized Gradient Approximation, GGA)Ҳ����Ϊ��һ�ַdz��õ�EXC[n(r)]���Ʊ���ʽ, ���������ڼ������ʵĽ����[14]��
��ʵ��Ӧ����, �ж����ֶο���ʵ�ֵ�һԭ���ļ���, Ŀǰ���㷺���õ���һ�ֳ�Ϊ����ƽ�沨(Pseudopotential Plane Wave)�ķ����� �÷����Ļ���˼���ǽ��ڲ���ӶԹ�������ɿ����Ƶ���������, �ȼ�Ϊһ���ڽ�ԭ�Ӻ�����ƽ���仯����Ч��, �����ݲ���ն���(Bloch��s Theorem)�������еĵ����Ӳ�����������ƽ�沨������չ����
2 ����������������ѡȡ
���о����õĵ�һԭ��������ͨ���ɽ��Ŵ�ѧ������Castep(Cambridge Sequential Total Energy Package)������[15]ʵ�ֵġ� ���Ӽ�������-�����ܷ���EXC[n(r)]����GGA�е�Perdew-Burke-Ernzerhof��ʽ[14]�� ѡȡ���ռ�����еij���(ultrasoft)����[16]��ȡ��ԭ�Ӻ����ڲ���Ӷ������ӵ������ơ� kֵȡ�����Monkhorst-Pack[17]����ѡȡ6��6��6����㡣 ƽ�沨����������Ŀ�ɶ��ܽضϵ�Ecut������, ��ѡȡ����Ek=(h2/2m)|k+G|2������Ecut��ƽ�沨��k(r)=ck+Gexp[i(k+G)��r]��Ϊչ�������е����Ӳ������Ļ������� ����֤ʵѡȡEcut=330.0eV���㹻���ܵ������� ����, �ڼ���ÿ��������ϵͳ�����Ļ�̬����֮ǰ, ���Ƕ���BFGS����[18]����Щ���������˼����Ż�, ��������ǵľ������ȶ��ṹ�� ��Ǣ����ʱ, ��ϵ������������ֵȡ5.0��10-7 eV, ÿ��ԭ���ϵ���Ҫ�����0.1eV/nm, ����ƫ��С��5.0��10-5 nm, Ӧ��ƫ��С��0.02GPa��
3 ������������
3.1 AlSb�Ľṹ�����ܷ���
��AlSb����п��ṹ�й������ּ�϶λ�ù������Ƕ��, ����ͼ1����ʾ��
��ͼ1�ɼ�, ��AlSb�ṹ���д����ʺ������Ƕ��ļ�϶λ��, ���ҵ��ռ��ȫ����϶λ�ú�, ���ɽ�һ��ȡ�����е�Alλ��, ����AlSb��Ƕ������Ƿdz��ɹ۵ġ�
ͼ2��ʾΪAlSb�������澧�����ı仯����ͼ�� ��(dEtot/da)|a0=0, �ɵ�AlSb��ƽ�⾧����a0=0.6084nm, ��ʵ��ֵaExp=0.6135nm�Ƚ��Ǻ�, ÿ��ԭ����Ӧ��ƽ������ΪE0tot=-846.7364eV��
��֪Al��ԭ�Ӱ뾶���۰뾶�ֱ�ΪraAl=0.1820nm��rcAl=0.1180nm; Sb��ԭ�Ӱ뾶���۰뾶�ֱ�ΪraSb=0.1530nm��rcSb=0.1400nm(�뾶����ȡ������CaRIne Crystallography 3.1)�� ��AlSb��, ���������Al-Sb��ԭ�Ӽ��ΪrAl-Sb=0.2631nm, ���ѷ�����
rcAl+rcSb��rAl-Si��raAl+raSb(4)
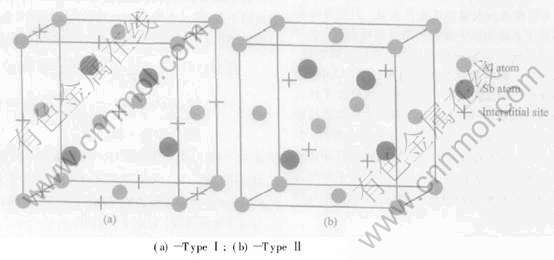
ͼ1 ��п��ṹAlSb�е����ּ�϶λ��
Fig.1 Two types of interstitial sites in AlSb with zinc-blende structure
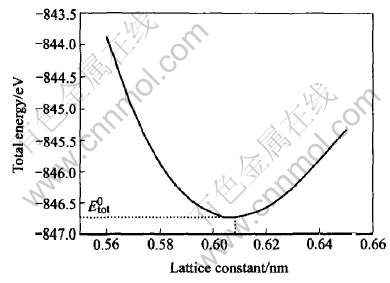
ͼ2 AlSb�����澧�����ı仯����
Fig.2 Curve of total energy of AlSb dependent on lattice constant
���AlSb��, ��ԭ�Ӽ�Ľ�ϼ��й����Բ������н����Բ��֡�
ͼ3��ʾΪAlSbԭ��(��һ��AlSb�����ӡ�)���ܴ��ṹͼ��̬�ܶ�ͼ�� ���ܴ��ṹͼ�п��Կ���AlSb�ļ۴��ܼ��뵼���ܼ�δ�����ܴ��ص�, ������ܼ�λ�ڴ�϶��, �۴���������ΪE+=0.005eV, ����������ΪE-=1.468eV, ���϶����ΪEg=E--E+=1.463eV, �ɴ˿�֪AlSbӦ����һ�ְ뵼����ϡ� ��̬�ܶ�ͼ�п��Կ����ڷ����渽����̬�ܶ�ֵN(EF)=0.229eV�dz�С, ����Ҫ��p̬����������, ����AlSb�Ľ����Էdz���, ���ӵĹ��л��̶Ƚϵ�, ���䵼�����ܲ��Ǻܺ�, �����ڽ�AlSb�Ƴɵ缫����ʱ, Ӧ���������ĵ���������ӵ缫�ĵ���������
3.2 AlSb��Ƕ������ܷ���
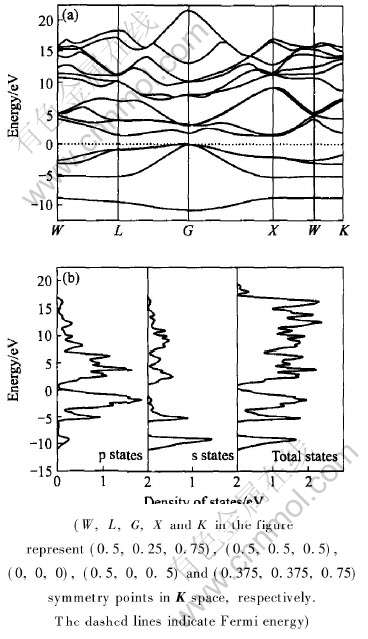
ͼ3 AlSbԭ����(a)�ܴ��ṹͼ��(b)̬�ܶ�ͼ
Fig.3 Band structure (a) and density of states (b) of AlSb primitive cell
����Li��Ƕ��AlSb�����в�δ�ı�AlSb�ĹǼܽṹ, ����Ƕ��ռ�ݵ�Type ���Type �����ּ�϶λ��, ��ôLiǶ��AlSb�ĵ绯ѧ��Ӧ�ɱ�ʾΪ
![]()
������ѧԭ����֪, ��Ӧ(5)ʽ�ļ���˹�����ܵı仯��rG(x)�뷴Ӧ��ƽ����ѹ��(x)�Ĺ�ϵΪ

�ּ���˹�����ܵı仯��rG(x)��дΪ

����ͨ�������P��V��T��S��ԶԶС�ڦ�E��, ��ʽ(7)�ɽ��Ʊ���Ϊ
![]()
����Ӧǰ����ϵ������֮�E�ֿɱ�ʾΪ

����ʽ(6)�� (8)��(9)�ɵ÷�Ӧʽ(5)��ƽ����ѹֵΪ

��֪Li������ΪEtot(Li)=-190.0273eV, AlSb������Etot(AlSb)=-211.6690eV�� Ϊ����Lix(AlSb)������, �ɹ��캬4��Alԭ�Ӻ�4��Sbԭ�ӵij���, ����һ����4��AlSb�����ӡ��ľ�������һ����������ѧ�еij���, Ȼ���ٽ�Li�����볬���е�Type ���Type ���϶λ��, �Եõ�����Li(AlSb)4�� Li2(AlSb)4�� Li3(AlSb)4�� Li4(AlSb)4�� Li5(AlSb)4�� Li6(AlSb)4�� Li7(AlSb)4�� Li8(AlSb)4�ij����ṹ, Ȼ���ټ�����Щ����������(���ھ���ͬһ�ַ���ʽLiz(AlSb)4�����м��ֲ�ͬLiռλ����ij���, ��ȡ�������ܵ���Сֵ)�� �ɴ˼��ɵ�Li1/4(AlSb)�� Li2/4(AlSb)�� Li3/4(AlSb)�� Li(AlSb)�� Li5/4(AlSb)�� Li6/4(AlSb)�� Li7/4(AlSb)�� Li2(AlSb)������, Ȼ���ٸ���ʽ(10)��ɼ�����γ�Lix(AlSb)���ƽ����ѹֵ, ���1��ʾ��
��1 Lix(AlSb)�����ӡ������ܼ��γ�Lix(AlSb)���ƽ����ѹֵ
Table 1 Total energies of Lix(AlSb) and average potentials for formations of Lix(AlSb) phases
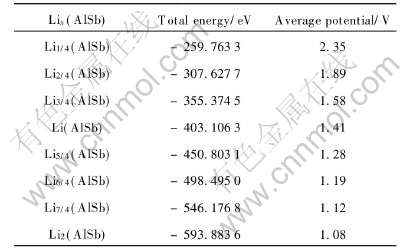
���ݱ�1�е�����, ����������Liռ��AlSb�еļ�϶λ��ʱ�ĵ�ѹ��Ƕ���������ͼ, ��ͼ4��ʾ��
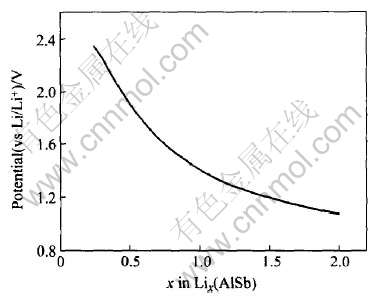
ͼ4 Liռ��AlSb�еļ�϶λ��ʱ�ĵ�ѹ����������
Fig.4 Curve of potential vs Li intercalated capacity for Li occupying interstitial sites in AlSb
��LiǶ����������, ��Liռ��AlSbȫ����϶λ�ú�, Li�ɽ�һ��ȡ��AlSb�ṹ�е�Alԭ��λ�����γɸ����Li3Sb�� �ڴ˹�����, ����Liȡ��Al���ı�AlSb�ľ���ṹ����, ��ֻʹ�þ���ĵ������������������ͻ���С�� ��ʱLi��Ƕ�뷴Ӧ�ɱ�Ϊ
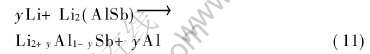
������ͬ���ķ����ɵ÷�Ӧʽ(11)��ƽ����ѹֵ��(x)Ϊ

��֪Al������ΪEtot(Al)=-57.2059eV, Li������ΪEtot(Li)=-190.0273eV, Li2(AlSb)������ΪEtot[Li2(AlSb)]=-593.8836eV�� ��ǰ��������������4�ֲ�ͬyֵ(y=0.25, 0.50, 0.75, 1)��Li2+yAl1-ySb������, ����ʽ(12)��ɼ�����γ�Li2+yAl1-ySb���ƽ����ѹֵ, ���2����ʾ��
��2 Li2+yAl1-ySb�����ܼ��γ�Li2+yAl1-ySb���ƽ����ѹֵ
Table 2 Total energies of Li2+yAl1-ySb and average potentials for formations of Li2+yAl1-ySb phases

�ɱ�2�е�����, ��������Liȡ��Al��ĵ�ѹ��Ƕ���������ͼ, ��ͼ5��ʾ��
���ͼ4��ͼ5�е�����, ��������ʽ
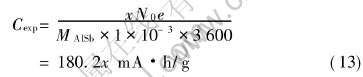
��Li��Ƕ��������ɵ�LiǶ��AlSb�缫ʱ
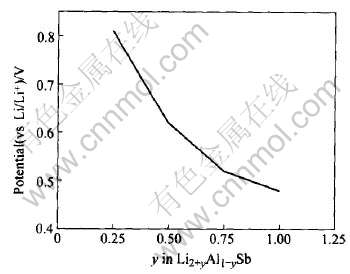
ͼ5 Liȡ��Al��ĵ�ѹ��Ƕ���������
Fig.5 Curve of potential vs Li intercalated capacity for Li substituting for Al
�������������� �ɴ˿�������LiǶ��AlSb�缫ʱ��ѹ����������������ͼ, ��ͼ6��ʾ�� ������Ҫָ������, ��ʽ(12)��, xΪǶ﮷�Ӧ�е�Ƕ﮸���, N0Ϊ�����ӵ�����, eΪ���ӵ���, MAlSbΪAlSb����Է���������
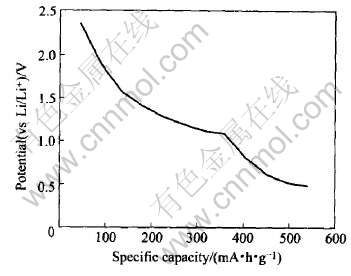
ͼ6 LiǶ��AlSb�缫ʱ�ĵ�ѹ����������������
Fig.6 Curve of potential vs specific capacity for Li intercalated into AlSb electrode
Honda��[19]���û�е�Ͻķ����ϳ���AlSb����Ʒ, ����������˵绯ѧ���ܵIJ���, �õ��˺������ŵ��µ�AlSb�缫�ĵ�ѹ����������������ͼ, ��֮��ͼ6�Աȷ��ֱȽ��Ǻ�, �Ӷ�֤ʵ�˱������߶�AlSb���ϵ�﮻���Ӧ���Ƶļ���, ��Li��Ƕ��AlSb����ʱ, ����ռ�ݵ������еļ�϶λ��, ���Ƕ����������, ��ռ����ȫ���ļ�϶λ�ú�, Li����ȡ��AlSb�е�Al�Ӷ��γɸ����Li3Sb��
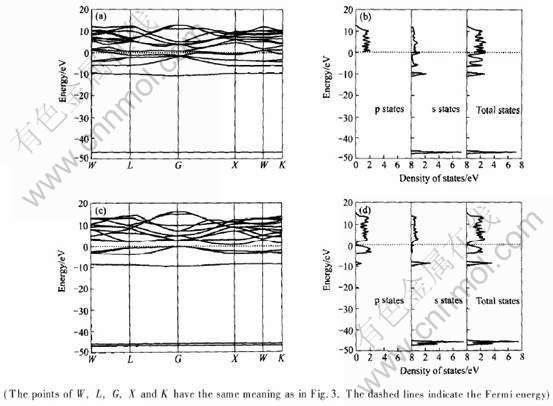
ͼ7 Li2AlSbԭ����(a)�ܴ��ṹͼ��(b)̬�ܶ�ͼ��Li3Sbԭ����(c)�ܴ��ṹͼ��(d)̬�ܶ�ͼ
Fig.7 Band structure (a) and density of states (b) of primitive cell of Li2AlSb and band structure (c) and density of states (d) of primitive cell Li3Sb
ͼ7���������ֵ��͵�Ƕ���Li2(AlSb)(ȡLix(AlSb)�е�x=2)��Li3Sb(ȡLi2+yAl1-ySb�е�y=1)���ܴ��ṹͼ��̬�ܶ�ͼ�� ��ͼ7(a)��, ���Կ���Li2(AlSb)�ļ۴��͵�����G���غ�, ����϶����Eg=0, ���ҷ����ܼ�EF�����뵼���С� �������LiǶ��AlSb�ļ�϶λ�ú��γɵļ�϶��������һ�ְ����, �䵼��������ԭ����AlSb������ߡ� ��ͼ7(b)��, ���Կ���Li2(AlSb)�ڷ����渽����̬�ܶ�ֵN(EF)=2.124eV, ������Ҫ��s̬��p̬���ӹ�ͬ��ɡ� �������Li2(AlSb)��, ����ӹ��л��̶��������, ������Ҳ��֮��ǿ�� ��ͼ7(c)��, Li3Sb�ļ۴��͵��������·ֿ�, ���϶����Eg=E--E+=0.753eV, �������ܼ�EF�������˴�϶�С� ��˵����Liռ��ȫ����϶λ�ú�, ��Liȡ��Al���γɵĹ������ֱ�Ϊ�뵼����, �䵼���������������͡� ��ͼ7(d)��, ���Կ���Li3Sb�ڷ����渽����̬�ܶ�ֵN(EF)=0.265 eV�� ��˵����Li3Sb�еĵ��ӹ��л��̶��������½�, �������Ҳ��֮�½��� Ϊ�˸������˵��AlSb�ĵ���������LiǶ���������ӵı仯���, ���ǽ���Ƕ���Lix(AlSb)(0��x��2)��Li2+yAl1-ySb(0��y��1)�Ĵ�϶����Eg���京����ı仯�������ͼ8�С� ��ͼ8��, ��϶����EgԽ���Ƕ���, �䵼������Խ��; ��֮, ��϶����EgԽС��Ƕ���, �䵼������Խ�á�
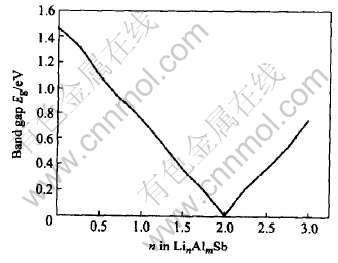
ͼ8 Ƕ���LinAlmSb�Ĵ�϶����Eg����Ƕ���n�ı仯����
Fig.8 Curve of band gap Eg of lithiated phases LinAlmSb dependent on its lithiated capacity n
4 ����
�ɵ�һԭ������ƽ�沨�������йص�����ѧԭ���õ��˵�LiǶ��AlSb�缫ʱ�����۵�ѹ����������������ͼ, �����ֵ�Li��Ƕ��AlSb�ľ���ṹʱ, ����ռ�ݵ������еļ�϶λ��, Ȼ�������Ƕ����������, ��ռ����ȫ����϶λ�ú�, Li���Խ�һ��ȡ��AlSb�е�Al�Ӷ��γɸ����Li3Sb�� ͨ������LiǶ��AlSbǰ����ܴ��ṹͼ��̬�ܶ�ͼ�ı仯, ����AlSb�ĵ�������������LiǶ���������Ӷ�����, ��Liռ��AlSb���м�϶λ��ʱ�ﵽ��ֵ, Li��һ�����Alʱ������������֮���͡�
REFERENCES
[1]Tarascon J M, Armand M. Issues and challenges facing rechargeable lithium batteries[J]. Nature, 2001, 414: 359-367.
[2]Santos Pe��a J, Brousse T, Schleich D M. Search for suitable matrix for the use of tin-based anodes in lithium ion batteries[J]. Solid State Ionics, 2000, 135(1-4): 87-93.
[3]Idota Y, Kubota T, Matsufuji A, et al. Tin-based amorphous oxide: a high-capacity lithium-ion-storage material[J]. Science, 1997, 276: 1395-1397.
[4]Massalski T B, Murray J L, Bennett L H, et al. Binary Alloy Phase Diagrams[M]. Ohio: America Society for Metals, Metals Park, 1987.
[5]Hamon Y, Brousse T, Jousse F, et al. Aluminum negative electrode in lithium ion batteries[J]. J Power Sources, 2001, 97-98: 185-187.
[6]Jeong G J, Kim Y U, Sohn H J, et al. Particulate-reinforced Al-based composite material for anode in lithium secondary batteries[J]. J Power Sources, 2001, 101: 201-205.
[7]Lindsay M J, Wang G X, Liu H K. Al-based anode materials for Li-ion batteries[J]. J Power Sources, 2003, 119-121: 84-87.
[8]л��, ���±�, �ܸ�ۿ, ��. �����仯����NiSb2����﮻���[J]. �й���ɫ����ѧ��, 2002, 12(6): 1238-1241.
XIE Jian, ZHAO Xin-bing, CAO Gao-shao, et al. Lithium absorption and release mechanism of intermetallic compound NiSb2[J]. The Chinese Journal of Nonferrous Metals, 2002, 12(6): 1238-1241.
[9]Ceder G, Chiang Y M, Sadoway D R, et al. Identification of cathode materials for lithium batteries guided by first-principles calculations[J]. Nature, 1998, 392: 694-696.
[10]Aydinol M K, Kohan A F, Ceder G, et al. Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides[J]. Phys Rev B, 1997, 56(3): 1354-1365.
[11]Ceder G, Aydinol M K, Kohan A F. Application of first-principles calculations to the design of rechargeable Li-batteries[J]. Comput Mater Sci, 1997, 8(1-2): 161-169.
[12]Hohenberg P, Kohn W. Inhomogeneous electron gas[J]. Phys Rev, 1964, 136(3B): B864-B871.
[13]Kohn W, Sham L J. Self-consistent equations including exchange and correlation effects[J]. Phys Rev, 1965, 140(4A): A1133-A1138.
[14]Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77(18): 3865-3868.
[15]Segall M D, Lindan P J D, Prober M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys Condens Matter, 2002, 14: 2717-2744.
[16]Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Phys Rev B, 1990, 41(11): 7892-7895.
[17]Monkhorst H J, Pack D. Special points for Brillouin-zone integrations[J]. Phys Rev B, 1976, 13(12): 5188-5192.
[18]Fischer T H, Almlof J. General methods for geometry and wavefunction optimization[J]. Journal of Physical Chemistry, 1992, 96: 9768-9774.
[19]Honda H, Sakaguchi H, Fukuda Y, et al. Anode behaviors of aluminum antimony synthesized by mechanical alloying for lithium secondary battery[J]. Mater Res Bull, 2003, 38(4): 647-656.
������Ŀ: ������Ȼ��ѧ����������Ŀ(50371103)
�ո�����: 2005-10-17; ������: 2006-01-13
ͨѶ����: ����, ����, ��ʿ; �绰: 0731-8830785; ����: 0731-8830785; E-mail: ycsu@mail.csu.edu.cn
