
J. Cent. South Univ. (2016) 23: 3239-3247
DOI: 10.1007/s11771-016-3389-1
Machine-learning-aided precise prediction of deletions with next-generation sequencing
GUAN Rui(����), GAO Jing-yang(�{����)
College of Information Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China
 Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Abstract:
When detecting deletions in complex human genomes, split-read approaches using short reads generated with next-generation sequencing still face the challenge that either false discovery rate is high, or sensitivity is low. To address the problem, an integrated strategy is proposed. It organically combines the fundamental theories of the three mainstream methods (read-pair approaches, split-read technologies and read-depth analysis) with modern machine learning algorithms, using the recipe of feature extraction as a bridge. Compared with the state-of-art split-read methods for deletion detection in both low and high sequence coverage, the machine-learning-aided strategy shows great ability in intelligently balancing sensitivity and false discovery rate and getting a both more sensitive and more precise call set at single-base-pair resolution. Thus, users do not need to rely on former experience to make an unnecessary trade-off beforehand and adjust parameters over and over again any more. It should be noted that modern machine learning models can play an important role in the field of structural variation prediction.
Key words:
1 Introduction
A deletion is a kind of genomic alteration that a segment of DNA is missing in comparison with a reference genome. As one of the most important classes of structural variations, deletions have broad effects on human complex diseases, physical traits, susceptibility to illness, and evolution [1]. For example, it has been found that GSTM1 homozygous deletion tends to increase bladder cancer risk [2], and 22q11.2 deletion is strongly associated with fetal conotruncal cardiac defects [3]. Inflammatory skin and bowel disease have been reported to be linked to ADAM17 deletion [4]. It is of great significance to sensitively detect deletions and precisely locate them at nucleotide resolution. Single-nucleotide breakpoint resolution is necessary for the study of variation formation mechanisms on one hand; on the other hand, it is the basis of genotyping and required to assess the functional impact of variations.
The emergence and development of next-generation sequencing has greatly promoted the change in the methods for discovering structural variations. A substantial number of sequencing-based computational approaches have recently sprung up [5]. These methods are divided into four major categories [6]: read-pair approaches [7], split-read technologies [8], read-depth analysis [9] and sequence assembly [10]. In essence, they all attempt to identify variant-related sequence features by mapping reads to the reference genome to finally call variations. However, the four types of strategy, on the other side, focus on different aspects when exploiting sequencing data, thus result in distinct abilities in the detection of structural variations. At present, split-read strategy is the most feasible one to detect deletions down to single-base-pair level [11].
Nevertheless, faced with the complex characteristics of human genomes, short reads generated using next- generation sequencing technologies, sequencing errors, low sequence coverage and the like obstacles, split-read approaches still have some challenges, one of which is that many false positives are called simultaneously. In order to tackle the problem, most of the current methods, such as Pindel [12], SRiC [13], SVseq [14-15], use a simple experiential threshold-based filtering strategy, that is, it winnows out a putative variation candidate if its certain supportive parameters values cannot meet the manually specified thresholds. Because of human experience limitation and inadequate utilization of variant-related sequence features, users adopting these approaches often have to make trade-off between sensitivity and false discovery rate (FDR). If expect to recall as many variations as possible, the number of false positives will generally increase; while if desire to obtain as precise call set as possible, more true variations will usually be missing.
Taking into account the influence of sequencing errors as well as sequence coverage and aiming at maximizing sensitivity while minimizing false discovery rate, we propose an integrated machine-learning-aided strategy for the detection of deletions. It organically combines the fundamental theories of read-pair approaches, split-read technologies and read-depth methods. Through analyzing initial mapping and split read alignment results, 16 deletion-related sequence features were extracted for the following machine- learning discrimination process to achieve the goal of obtaining a both sensitive and precise call set at single- base-pair resolution.
2 Methods
2.1 Features
According to the rationale behind the three mainstream methods, read-pair approaches, split-read technologies and read-depth methods, we extracted 16 deletion-related sequence features. These features are listed in Table 1.
2.1.1 Discordant read pairs
A mapped read pair with concordant order and orientation but a too large mapping span usually indicates a deletion in the corresponding genomic region. Under normal circumstances, a read pair that spans a deletion has both ends mapped to the reference properly, but the mapping span is greater than the library insert size (Fig. 1(a)). Another situation is that one end of a read pair happens to harbor the joint point brought by a deletion event and the end is left soft clipped as a result, while the other end is well mapped as a whole too far apart (Fig. 1(b)).
We computed discordant read pairs from BAM files. A read pair with mapping span longer than ��+3�� is considered discordant in our method. Here, �� is the mean library insert size and �� is its standard deviation. These two variables can be either specified by user or calculated from the given BAM file. As mentioned before, we care about two sorts of read pairs with discordant mapping spans. One is that has two ends properly mapped, that is, the CIGAR fields of the both ends are ��100M�� when read length is 100bp for example. The other is with one end ��100M��, while the other end may be ��15S85M��. The total number of these two kinds of discordant read pairs is recorded as the first group of features, which is called dis_cnt.
2.1.2 Split mapping
After initial mapping, in addition to the read pairs that have both ends mapped to the reference genome, there are still a number of read pairs with one end mapped while the other not perfectly mapped (like unmapped, gaped aligned, with too many mismatches and so on), which is often due to being sampled across a variation breakpoint (Fig. 2). As in a case of deletion, if break the read into two parts in an appropriate way and realign them separately in the genomic region determined by its mate (called anchor), the exact breakpoints and the deleted segment compared to the reference should be computed.
Table 1 Deletion-related sequence features
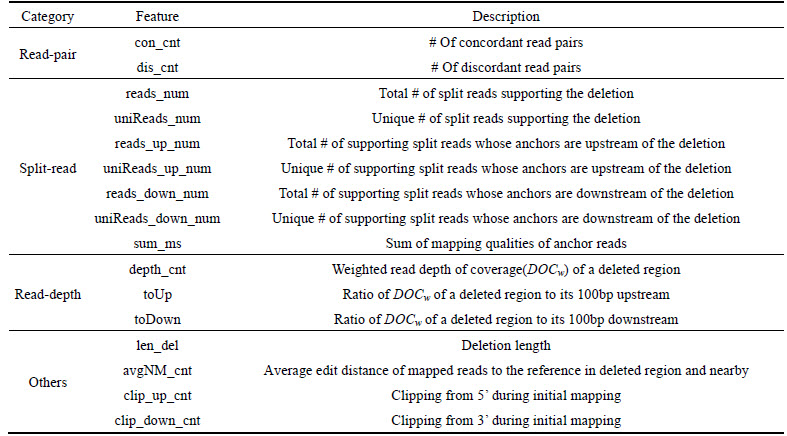
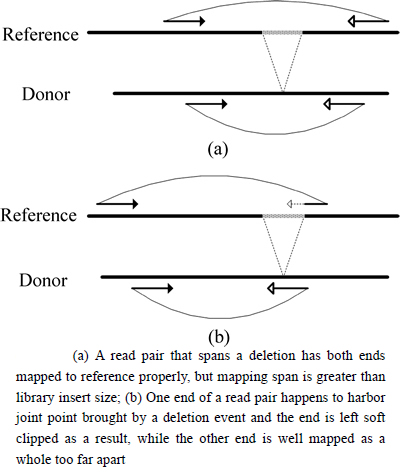
Fig. 1 Discordant mapping span of read pairs harboring a deletion:
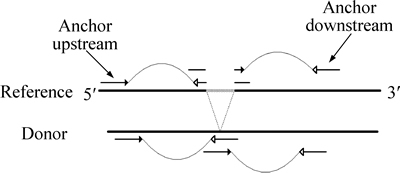
Fig. 2 Split alignments of read pairs (A read sampled across a deletion breakpoint can be broken into two parts in an appropriate way and have them realigned separately in the genomic region determined by its mate, the so-called anchor)
To illustrate the integrated idea that we come up with in a concrete way, we used Pindel as split mapping tool in the following experiment. Here, our one and only goal is to make Pindel call deletions as many as possible, regardless of false positives, for those false positive ones will finally be filtered out in our following machine- learning discrimination process. Thus, we set the sensitivity parameter (-E) of Pindel to 0.99 and the minimum support for event (-M) to 2. Considering the position-dependency structure of Illumina read quality profiles and dissimilar error profiles between the two ends of read pairs, seven split-mapping features are selected for deletions based on Pindel alignment results: total number of reads and unique number of reads supporting the deletion(reads_num, uniReads_num), total number of supporting reads and unique number of supporting reads whose anchors are upstream of the deletion(reads_up_num, uniReads_up_num), total number of supporting reads and unique number of supporting reads whose anchors are downstream of the deletion(reads_down_num, uniReads_down_num), and sum of mapping qualities of anchor reads(sum_ms). When in practice, other split mapping algorithms can also be applied, but need to follow the principle: to recall as many variants as possible and to extract the seven features mentioned above.
2.1.3 Read depth of coverage
Assuming that the sequencing process is uniform, read depth follows a random (typically Poisson or modified Poisson) distribution and the number of reads mapping to a genomic region is expected to be proportional to the number of times that the region appears in the sequenced sample. Hence, there will be zero or a lower number of reads mapping to a deleted region compared with the regions up- and downstream thereof (Fig. 3).
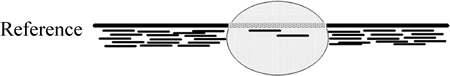
Fig. 3 Illustration of read depth of coverage in a deleted region and its adjacent regions (There will be zero or a lower number of reads mapping to a deleted region compared with the regions up- and downstream thereof)
Taking mapping quality into consideration, the weighted read depth of coverage DOCw is calculated by
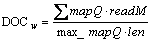 (1)
(1)
where readM counts the number of alignment matches of a read that maps to a particular region of length len with mapping quality mapQ scaled by the overall maximum mapping quality max_mapQ. DOCw of a region preliminarily inferred to be deleted(depth_cnt) and its ratios to DOCw of 100bp up- and downstream of the region(toUp, toDown) respectively are taken as the third group of features.
2.1.4 Others
Information like ��S�� (soft clipping), ��D�� (deletion from the reference) and so on in initial mapping results, which is often ignored by most structural variation discovery methods, is useful indeed. It usually indicates the exact breakpoint of a potential variation (Fig. 4). Given a candidate deletion, we counted the number of reads that clipped or partially deleted at the exact breakpoint loci. Because of different sequencing quality between 5�� end and 3�� end of a read, we separated the two situations (that is, clipping from 5�� during initial mapping and from 3��) and computed clip_up_cnt and clip_down_cnt respectively.
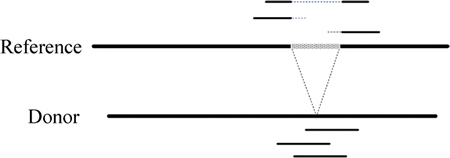
Fig. 4 Split alignment results offered by initial mapping algorithms (Information like ��S�� (soft clipping), ��D�� (deletion from reference) and so on in initial mapping results usually indicates the exact breakpoint of a potential variation)
Moreover, the farther the edit distance of a mapped read to the reference, the less reliable the mapping result. Thus, given a deletion candidate, we collected all the reads that mapped to the reference in the inferred deleted region and ��+3�� base pairs up- and down-stream of the region from BAM files, and calculated the average edit distance avgNM_cnt. We also took deletion length into consideration, for importance of signatures discussed above would change with deletion length. For example, the shorter the deletion candidate is, the more reliable the split-read signatures are, and read-pair signatures would be more reliable when candidate event is longer.
2.2 Machine learning algorithms
2.2.1 Support vector machine
Support vector machine (SVM) is a popular machine learning method for classification based on statistical learning theory and has been widely used in many fields [16]. It uses a nonlinear mapping to transform the original training data into a sufficiently higher dimensional space, within which, it searches for the linear optimal separating hyperplane to separate data from two classes. Given training vectors xi��Rn, i=1, ��, l, in two classes, and an indicator vector y��Rl such that yi��{+1, -1}, it solves the following primal optimization problem [17]:
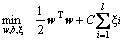
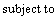
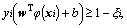
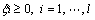 (2)
(2)
where ��(xi) maps xi into a higher dimension and C>0 is the regularization parameter. SVM is highly accurate and much less prone to overfitting than other methods.
2.2.2 AdaBoost
AdaBoost [18-19] (short for adaptive boosting) is the most important variant of boosting algorithm which is an ensemble method for combining a series of weighted base classifiers. It adaptively changes distribution of training data by focusing more on previously misclassified samples, that is, AdaBoost initially assigns each training sample, an equal weight, and as the training continues, weights of samples misclassified by the base classifier will be increased and samples that are classified correctly will have their weights decrease. Given a data set of N tuples (x, y), where yi is the class label of xi (i=1, ��, N), generating T classifiers C for the ensemble requires T to round through the algorithm. Importance of a base classifier is calculated by
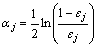 (3)
(3)
where ��j is error rate of base classifier Cj:
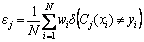 (4)
(4)
and weight of each sample is updated according to
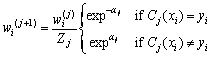 (5)
(5)
where Zj is the normalization factor. If any intermediate rounds to produce error rate higher than 50%, the weights are reverted back to 1/N and the resampling procedure is repeated. Class prediction is made by
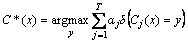 (6)
(6)
AdaBoost can significantly improve accuracy in comparison to a single individual.
2.2.3 Random forests
Random forest [20] is a collection of decision tree classifiers that are generated using a random selection of attributes at each node to determine the split. More formally, each tree depends on the values of a random vector sampled independently and with the same distribution for all trees in the forest. During classification, each tree votes and the most popular class is returned. Random forests are comparable in accuracy to AdaBoost and sometimes better, yet are more robust to errors and outliers. Besides, the method gives useful internal estimates of error, strength, correlation and variable importance, which is especially important to the study of life sciences.
3 Results and analysis
3.1 Simulated data
To evaluate performance of our method, we implanted 792 deletions identified on chromosome 20 of Craig Venter��s genome [21] into the NCBI build 36 human reference. Both homozygous and heterozygous deletions were introduced, and the heterozygous ones were placed according to the corresponding haplotype file. We used ART [22], a next-generation sequencing read simulator, to simulate five sets (coverage 5x, 10x, 20x, 30x, 40x) of 75bp Illumina [23] paired-end reads with insert size of a 500bp mean (length of sequence between the paired-end adapters) and a 10bp standard deviation based on the built-in Illumina-specific read error model (��0.03) and empirical read quality score distribution.
3.2 Performance assessment
BWA-MEM [24] algorithm was used to initially map the simulated paired-end reads to the reference and Pindel, a pattern-growth split-read approach, was then applied to forming a candidate deletion set. Considering sensitivity only (i.e. false discovery rate is not controlled at the same time), to recall as many deletions as possible, we set the sensitivity parameter (-E) of Pindel to 0.99 and the minimum support for event (-M) to 2. Candidate results are summarized in Table 2.
Table 2 Statistics of deletion candidates
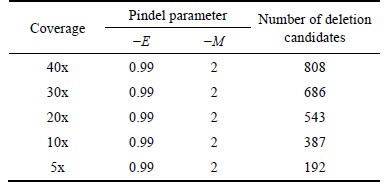
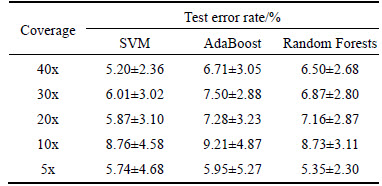
On the basis of both the initial mapping and the following split read alignment results, 16 features mentioned previously were extracted for each deletion candidate. A certain number of deletion candidates that had been labeled as true or false after being compared with the 792 simulated benchmark deletions were randomly selected from the candidate set, and were used to train a machine learning model. To illustrate effectiveness and necessity of machine learning models on sensitivity maximization and false discovery rate minimization, we tested three modern machine learning algorithms, SVM, AdaBoost neural net ensemble (one- hidden-layer backpropagation neural networks adopted as the base classifiers) and Random Forests, in the experiment. Since split-read algorithms like Pindel pinpoint the breakpoint of the variation with single-base- pair resolution, we take a strict criterion to evaluate deletion calls, that is, we consider a predicted deletion correct if and only if its start and length are identical to those of a simulated benchmark deletion. We performed a 10-fold cross-validation and repeated each cross- validation 10 times. Table 3 lists test error of the SVM using a radial basis kernel, the 20-week-learning- BP AdaBoost ensemble network and the Random Forests with 20 decision trees.
The trained models were then applied to the five deletion candidate sets. Corresponding results are recorded in Table 4. For each depth of coverage, five classes of methods are analyzed: P1 directly calls deletions using Pindel with the sensitivity parameter (-E) set to 0.99 and the minimum support for event (-M) set to 2, SVM-aided S, AdaBoost-aided A, Random-Forests- aided R and P2 directly calls deletions using Pindel with the sensitivity parameter (-E) set to 0.90 and the minimum support for event (-M) set to 3. Performances of the five methods are compared and demonstrated in Fig. 5.
Table 3 Test error of SVM, AdaBoost ensemble network and Random Forests
Aided by machine learning models that are trained on deletion-related features extracted from different aspects according to diverse theories, it is able to effectively reduce the number of false positives with ignorable loss of sensitivity. This is especially noticeable when read depth of coverage is relatively high. For instance, for coverage 20x, 30x and 40x respectively, true positive rates (TPR) of the five methods are comparative, while S, A and R methods that introduce machine learning models have obviously lower false discovery rate, such as A method down to 5.68%, 6.69% and 6.99% in turn.
When read depth of coverage is lower, S, A and R methods that are combined with machine learning algorithms can ensure lower false discovery rate while maintaining higher true positive rate. For example, for coverage 5x and 10x, true positive rates of S, A and R methods are much higher than P2, such as R method increased by 10.10% and 7.83% separately, and false discovery rates of them are all lower than P1.
Figures after symbol ��|�� in column ��FP�� of Table 4 are numbers of false positives that overlap with the simulated benchmark deletions. Further analysis on the false positive calls of S, A and R methods reveals that over 66.7% of the false positive calls overlap with the simulated benchmark deletions and this may be one of the main causes of them not being distinguished by machine learning models.
Comparison of the three methods S, A and R suggests that, no matter for high or low read depth of coverage, R method always has the lowest false discovery rate, while true positive rate of S method is slightly higher than that of the other two methods.
Table 4 Results of deletion-detection experiment
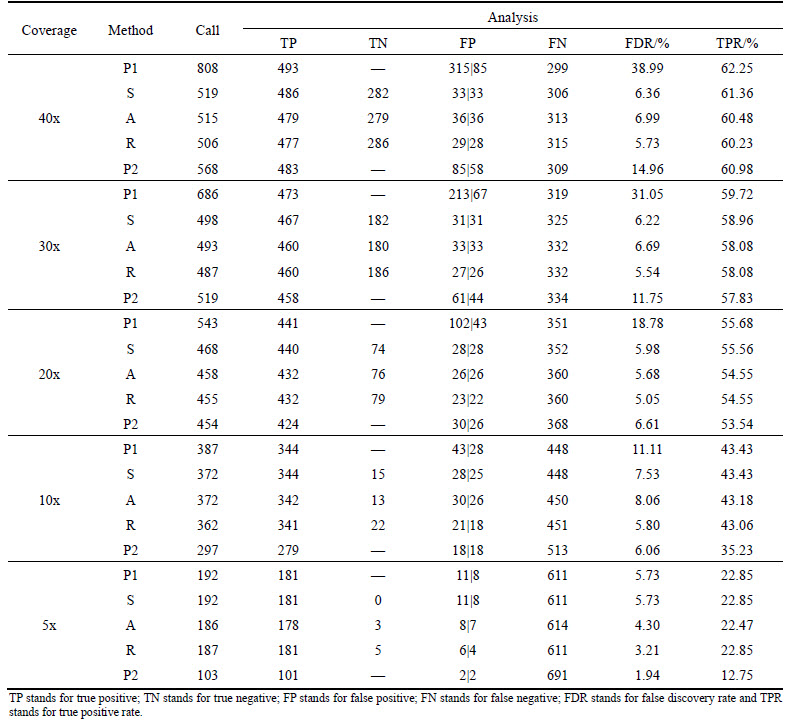
Moreover, many deletions in the simulated benchmark set occur in repetitive regions that are difficult to map using short reads generated from next-generation sequencing platform. This may be a reason why even when coverage is as high as 40x. There are still quite a number of deletions (��38%) not being detected.
3.3 Feature analysis
If a feature plays an important role in a random forest decision process, then when we add some noise to the feature, that is, to change the value of the feature randomly, prediction accuracy of the random forest will decrease significantly. Based on such heuristic rules, random forest itself can depict importance of each variable (i.e. permutation importance). We made variable importance internal evaluation of the 16 features described in section 2.1 with the R model constructed in section 3.2, and results are shown in Fig. 6.
When read depth of coverage is low, the number of discordant read pairs and split reads is reduced, and sometimes, we may not even obtain the two types of sequence features for some deletion events. Therefore, DOCw of a region preliminarily inferred to be deleted and its ratios to DOCw of 100bp up- and downstream of the region respectively are more important than the number of discordant read pairs and split reads when coverage is low. In addition, compared to high-coverage circumstance, the impact of the three features, deletion length, the number of concordant read pairs and the average edit distance of mapped reads to the reference in deleted region and nearby, on discriminant model prediction accuracy is bigger when coverage is low.
As the read depth of coverage increases, so does the number of discordant read pairs and split reads that support related deletion events. And these features become more and more important. When coverage is higher, the importance of the number of split reads supporting the deletion and sum of mapping qualities of anchor reads goes beyond DOCw of a inferred deleted region and its ratios to DOCw of 100bp up- and downstream of the region respectively. For paired-end reads generated from Illumina, the error profiles between the first and the second reads are significantly different. The error rate of the first reads is lower than that of the second reads. So, breaking the first reads into two shorter parts to do alignment usually gets more credible results. Hence, the number of supporting split reads whose anchors are downstream of a deletion has more influence on prediction accuracy than that of supporting split reads whose anchors are upstream of the deletion. Moreover, because the higher the coverage is, the more the reads are, information like ��S��, ��D�� and so on in initial mapping results grows and plays an increasingly important role in decision process.
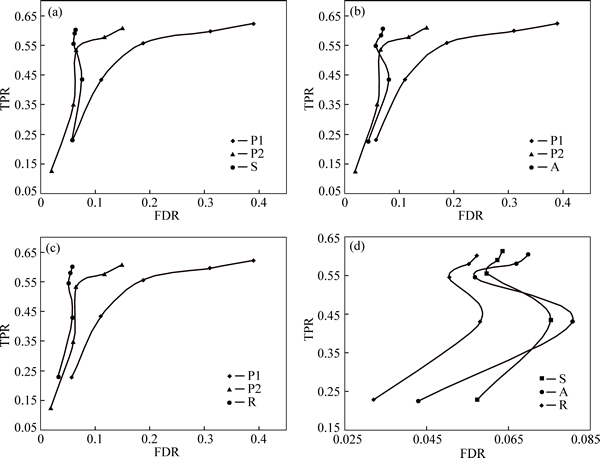
Fig. 5 Performances of five methods for deletion prediction (Five methods are divided into four groups for comparison. First, each machine-learning-aided method alone is compared with P1 and P2 in terms of true positive rate and false discovery rate. Then comparison of three methods S, A and R is carried out)
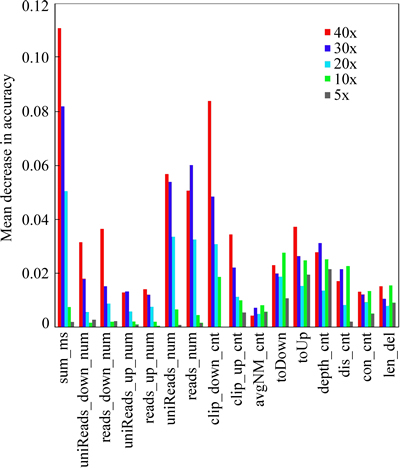
Fig. 6 Mean decrease in accuracy of R prediction model
4 Conclusions
1) Higher sensitivity and lower false discovery rate can be ensured at the same time by adopting the integrated strategy. The strategy organically combines the fundamental theories of the three mainstream methods (read-pair approaches, split-read technologies and read-depth analysis) with machine learning algorithms to obtain a sensitive and precise call set at single-base-pair resolution. The first step, regardless of false discovery rate, attempts to find deletions with exact breakpoints as many as possible (i.e. to maximize sensitivity) based on split read alignment following initial mapping. The second step, for each deletion found in the first step (so-called candidate), extracts the discussed 16 deletion-related features according to the rationale behind the three mainstream methods. The third step applies a modern machine learning model to distinguishing true from false deletion candidates (i.e. to minimize false discovery rate).
2) The machine-learning-aided strategy can effectively reduce the number of false positives with negligible loss of sensitivity. Therefore, users do not need to rely on former experience to make an unnecessary trade-off between sensitivity and false discovery rate beforehand any more, and as well no longer need to adjust parameters over and over again. It should be noted that modern machine learning models can be a real boon in detection of structural variations if being introduced appropriately.
References
[1] MCVEAN G A, ALTSHULER D M, DURBIN R M, ABECASIS G R, BENTLEY D R, CHAKRAVARTI A, CLARK A G, DONNELLY P, EICHLER E E, FLICEK P, GABRIEL S B, GIBBS R A, GREEN E D, HURLES M E, KNOPPERS B M, KORBEL J O, LANDER E S, LEE C, LEHRACH H, MARDIS E R, MARTH G T, MCVEAN G A, NICKERSON D A, SCHMIDT J P, SHERRY S T, WANG J, WILSON R K, GIBBS R A, DINH H, KOVAR C, et al. An integrated map of genetic variation from 1,092 human genomes [J]. Nature, 2012, 491(7422): 56-65.
[2] MOORE L E, BARIS D R, FIGUEROA J D, GARCIA-CLOSAS M, KARAGAS M R, SCHWENN M R, JOHNSON A T, LUBIN J H, HEIN D W, DAGNALL C L, COLT J S, KIDA M, JONES M A, SCHNED A R, CHERALA S S, CHANOCK S J, CANTOR K P, SILVERMAN D T, ROTHMAN N. GSTM1 null and NAT2 slow acetylation genotypes, smoking intensity and bladder cancer risk: results from the New England bladder cancer study and NAT2 meta-analysis [J]. Carcinogenesis, 2011, 32(2): 182-189.
[3] LEE M Y, WON H S, BAEK J W, CHO J H, SHIM J Y, LEE P R, KIM A. Variety of prenatally diagnosed congenital heart disease in 22q11.2 deletion syndrome [J]. Obstetrics & Gynecology Science, 2014, 57(1): 11-16.
[4] BLAYDON D C, BIANCHERI P, DI W L, PLAGNOL V, CABRAL R M, BROOKE M A, VAN HEEL D A, RUSCHENDORF F, TOYNBEE M, WALNE A, O��TOOLE E A, MARTIN J E, LINDLEY K, VULLIAMY T, ABRAMS D J, MACDONALD T T, HARPER J I, KELSELL D P. Inflammatory skin and bowel disease linked to ADAM17 deletion [J]. New England Journal of Medicine, 2011, 365(16): 1502-1508.
[5] MILLS R E, WALTER K, STEWART C, HANDSAKER R E, CHEN K, ALKAN C, ABYZOV A, YOON S C, YE K, CHEETHAM R K, CHINWALLA A, CONRAD D F, FU Y, GRUBERT F, HAJIRASOULIHA I, HORMOZDIARI F, IAKOUCHEVA L M, IQBAL Z, KANG S, KIDD J M, KONKEL M K, KORN J, KHURANA E, KURAL D, LAM H Y K, LENG J, LI R, LI Y, LIN C-Y, LUO R, et al. Mapping copy number variation by population-scale genome sequencing [J]. Nature, 2011, 470(7332): 59-65.
[6] ALKAN C, COE B P, EICHLER E E. Genome structural variation discovery and genotyping [J]. Nature Reviews Genetics, 2011, 12(5): 363-376.
[7] BUCZKOWICZ P, HOEMAN C, RAKOPOULOS P, PAJOVIC S, LETOURNEAU L, DZAMBA M, MORRISON A, LEWIS P, BOUFFET E, BARTELS U, ZUCCARO J, AGNIHOTRI S, RYALL S, BARSZCZYK M, CHORNENKYY Y, BOURGEY M, BOURQUE G, MONTPETIT A, CORDERO F, CASTELO-BRANCO P, MANGEREL J, TABORI U, HO K C, HUANG A, TAYLOR K R, MACKAY A, BENDEL A E, NAZARIAN J, FANGUSARO J R, KARAJANNIS M A, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations [J]. Nature Genetics, 2014, 46(5): 451-456.
[8] MARSCHALL T, HAJIRASOULIHA I,  A. MATE-CLEVER: Mendelian-inheritance-aware discovery and genotyping of midsize and long indels [J]. Bioinformatics (Oxford, England), 2013, 29(24): 3143-3150.
A. MATE-CLEVER: Mendelian-inheritance-aware discovery and genotyping of midsize and long indels [J]. Bioinformatics (Oxford, England), 2013, 29(24): 3143-3150.
[9] ABYZOV A, URBAN A E, SNYDER M, GERSTEIN M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing [J]. Genome Research, 2011, 21(6): 974-984.
[10] GNERRE S, MACCALLUM I, PRZYBYLSKI D, RIBEIRO F J, BURTON J N, WALKER B J, SHARPE T, HALL G, SHEA T P, SYKES S, BERLIN A M, AIRD D, COSTELLO M, DAZA R, WILLIAMS L, NICOL R, GNIRKE A, NUSBAUM C, LANDER E S, JAFFE D B. High-quality draft assemblies of mammalian genomes from massively parallel sequence data [J]. Proceedings of the National Academy of Sciences of the United States of America, 2011, 108(4): 1513-1518.
[11] PAVLOPOULOS G A, OULAS A, IACUCCI E, SIFRIM A, MOREAU Y, SCHNEIDER R, AERTS J, ILIOPOULOS I. Unraveling genomic variation from next generation sequencing data [J]. BioData Mining, 2013, 6(1): 1-25.
[12] YE Kai, SCHULZ M H, LONG Quan, APWEILER R, NING Ze-min. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads [J]. Bioinformatics, 2009, 25(21): 2865-2871.
[13] ZHANG Zheng-dong, DU Jiang, LAM H, ABYZOV A, URBAN A E, SNYDER M, GERSTEIN M. Identification of genomic indels and structural variations using split reads [J]. BMC Genomics, 2011, 12(1): 1-12.
[14] ZHANG Jin, WU Yu-feng. SVseq: An approach for detecting exact breakpoints of deletions with low-coverage sequence data [J]. Bioinformatics, 2011, 27(23): 3228-3234.
[15] ZHANG Jin, WANG Jia-yin, WU Yu-feng. An improved approach for accurate and efficient calling of structural variations with low-coverage sequence data [J]. BMC bioinformatics, 2012, 13 (Suppl 6): 1-11.
[16] LIU Qin, PENG Ke, LIU Wei, XIE Qin, LI Zhong-yang, LAN Hao, JIN Yao. Fingerprint singular points extraction based on orientation tensor field and Laurent series [J]. Journal of Central South University, 2014, 21(5): 1927-1934.
[17] CORTES C, VAPNIK V. Support-vector networks [J]. Machine Learning, 1995, 20(3): 273-297.
[18] FREUND Y, SCHAPIRE R E. A decision-theoretic generalization of on-line learning and an application to boosting [J]. Journal of Computer and System Sciences, 1997, 55(1): 119-139.
[19] GAO Jing-yang, CHEN Cheng-li-zhao, ZHU Qun-xiong. A new variant Boosting algorithm: Update sample��s weight according to standard deviation of Error-Right statistics [J]. Journal of Central South University: Science and Technology, 2012, 43(11): 4355- 4360. (in Chinese)
[20] BREIMAN L. Random forests [J]. Machine Learning, 2001, 45(1): 5-32.
[21] LEVY S, SUTTON G, NG P C, FEUK L, HALPERN A L, WALENZ B P, AXELROD N, HUANG J, KIRKNESS E F, DENISOV G, LIN Y, MACDONALD J R, PANG A W C, SHAGO M, STOCKWELL T B, TSIAMOURI A, BAFNA V, BANSAL V, KRAVITZ S A, BUSAM D A, BEESON K Y, MCINTOSH T C, REMINGTON K A, ABRIL J F, GILL J, BORMAN J, ROGERS Y H, FRAZIER M E, SCHERER S W, STRAUSBERG R L, et al. The diploid genome sequence of an individual human [J]. PLoS biology, 2007, 5(10): e254.
[22] HUANG Wei-chun, LI Le-ping, MYERS J R, MARTH G T. ART: A next-generation sequencing read simulator [J]. Bioinformatics, 2012, 28(4): 593-594.
[23] LAM H Y K, CLARK M J, CHEN Rui, CHEN Rong, NATSOULIS G, O��HUALLACHAIN M, DEWEY F E, HABEGGER L, ASHLEY E A, GERSTEIN M B, BUTTE A J, JI H P, SNYDER M. Performance comparison of whole-genome sequencing platforms [J]. Nature Biotechnology, 2011, 30(1): 78-82.
[24] LI Heng. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM [J]. arXiv:1303.3997 [q-bio], 2013.
(Edited by YANG Hua)
Foundation item: Project(61472026) supported by the National Natural Science Foundation of China; Project(2014J410081) supported by Guangzhou Scientific Research Program, China
Received date: 2016-01-20; Accepted date: 2016-05-05
Corresponding author: GAO Jing-yang, PhD, Professor; E-mail: gaojy@mail.buct.edu.cn
Abstract: When detecting deletions in complex human genomes, split-read approaches using short reads generated with next-generation sequencing still face the challenge that either false discovery rate is high, or sensitivity is low. To address the problem, an integrated strategy is proposed. It organically combines the fundamental theories of the three mainstream methods (read-pair approaches, split-read technologies and read-depth analysis) with modern machine learning algorithms, using the recipe of feature extraction as a bridge. Compared with the state-of-art split-read methods for deletion detection in both low and high sequence coverage, the machine-learning-aided strategy shows great ability in intelligently balancing sensitivity and false discovery rate and getting a both more sensitive and more precise call set at single-base-pair resolution. Thus, users do not need to rely on former experience to make an unnecessary trade-off beforehand and adjust parameters over and over again any more. It should be noted that modern machine learning models can play an important role in the field of structural variation prediction.
