

Molecular dynamics simulation of ion selectivity traits of nickel hexacyanoferrate thin films
HAO Xiao-gang(������)1, YU Qiu-ming2, JIANG Shao-yi2, D. T. SCHWARTZ2
1. Department of Chemical Engineering, Taiyuan University of Technology, Taiyuan 030024, China;
2. Department of Chemical Engineering, University of Washington, Seattle, Washington 98195-1750, USA
Received 9 August 2005; accepted 19 January 2006
Abstract:
The ion selectivity of nickel hexacyanoferrate thin film to alkali cations in ESIX (electrochemically switched ion exchange) processes was investigated using molecular dynamics(MD) techniques; water and cation (Na+ and Cs+) intercalation, configuration, and dynamics in reduced nickel hexacyanoferrate structures with different cation combinations were studied and compared with the experimental results. In the simulations, water was represented by an extended simple point-charge(SPC/E) model, and all other atomic interactions were represented by a universal force field(UFF). The potential energies of various cations combination (Cs+ and Na+) in reduced i-NiHCF and 1 mol/L Cs/NaCl mixed solution were obtained. In most cases, the total potential energy of the solid is reduced when water is intercalated into the various reduced NiHCF structures. Combining the solid and the solution simulation results, it is shown that the solid composition of 3Cs+/1Na+ is the stablest structure form (NaCs3Ni4[Fe(CN)6]3) over a range of solution compositions.
Key words:
nickel hexacyanoferrate; molecular dynamics simulation; ion selectivity; thin film; electrochemically switched ion exchange;
1 Introduction
Nickel hexacyanoferrate(NiHCF) is a zeolite-like Prussian blue analogue, where cyanide bonding between nickel and iron creates a cubic unit cell with intercalated cations that balance the net negative charge on the matrix. When NiHCF is grown as a thin film on conductive substrate[1-5], it can be electrochemically switched between reduced and oxidized states, reversibly intercalating and deintercalating alkali cations from solution[6,7], realizing the electrochemically switched ion exchange(ESIX) process. The intercalation selectivity for electrochemically prepared films and the relationship between the structure and ion intercalation have been measured quantitatively using different methods[6-9]. The cathodically deposited NiHCF thin films intercalate different proportions of Cs+ and Na+ (or K+) depending on both solution concentration and the charge density on the matrix[7,10,11]. Cs+/Na+ (or K+) partitioning in oxidized NiHCF matrices (i.e. lower charge densities) displays a high Cs+ selectivity for all solution compositions studied. In contrast, Cs+/Na+ (or K+) partitioning in the more highly charged reduced matrix exhibits a reversal of selectivity, going from a Cs+-selective state when the solution mole fraction (x(Cs)) is below 0.7 to a K+-selective state when x(Cs)��0.7[7,10]. But the selectivity mechanism of the NiHCF thin film to Cs+/K+ is not clear.
The number of water molecules and cations intercalated in NiHCF depends strongly on the structure and oxidation state of the material[12]. Molecular simulation methods have been used to study the characteristics of water and ion intercalation into the three different structural forms of NiHCF[13]��s-NiHCF, i-NiHCF and d-NiHCF. Simulation results show that water and ion intercalation behavior is sensitive to the matrix structure and the i-NiHCF is the primary structure of electrodeposited film[12].
In order to elucidate the ion selectivity traits of the NiHCF thin film to Cs+/Na+, the authors use molecular dynamics(MD) simulation to study the characteristics of water and ion intercalation into various reduced i-NiHCF matrices with different ion-combination in this paper.
The primary focus is on the total potential energy differences of NiHCF solid containing Cs+ and/or Na+ when extra waters (beyond the coordination sphere waters) are added to the reduced i-NiHCF matrices. Combining the aqueous solution (Na/CsCl) simulation results, the stable film composition of reduced NiHCF matrices can be confirmed when different combinations of alkali cations are intercalated into the solid.
2 Molecular dynamics simulation
All the simulations were performed using the materials simulation software package, Cerius2, from Molecular Simulations Inc. (San Diego, CA, USA).
2.1 Solid structure of i-NiHCF
The geometry and charge distribution for various reduced NiHCF were determined using the periodic DMol3 density functional theory (DFT) software module. The structural form of reduced i-NiHCF with intercalated Na+ is shown in Fig.1. The six added waters in i-NiHCF satisfy the octahedral coordination sphere for lattice Ni+2 and the connection between the nickel and oxygen atoms is a guide for the eye.

Fig.1 Unit cell structural form of i-NiHCF in reduced state Na4Ni4[Fe(CN)6]3��6H2O
2.2 Force fields and charges
The charges on solid matrix atoms and intercalated cations were calculated using the charge equilibration (QEq) method[14]. Once the charges were determined, they were fixed during subsequent MD simulations. The electrostatic interactions were described using the constant-epsilon functional form:
![]() (1)
(1)
where C=1 388.026 3 converts E to units of kJ/mol when qi and qj are given in electron units. Ewald sums were used to deal with the long-range electrostatic interactions.
In these simulations, the atoms of the NiHCF matrix were fixed at the equilibrium positions obtained from nonlocal DFT calculations. Thus, interactions between atoms within the solid matrix were neglected but all other interactions were considered. The total force fields included water-water, water-alkali metal cation, and cation-cation interactions as well as water- and cation-solid matrix atom interactions. The bond stretching and bending as well as Van der Waals and Coulombic interactions in the system was described by the universal force field(UFF)[15]. The extended simple point charge(SPC/E) model[16] was chosen to represent water molecules. Sodium and cesium cations, as well as all the solid matrix atoms, were represented by point charges having Lennard-Jones(LJ) centers on them. The LJ 12-6 potential model in Cerius is written as
![]() (2)
(2)
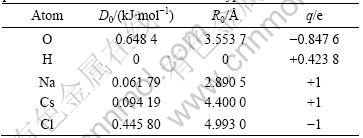
The parameters D0 and R0 for all solid matrix atoms were adopted from the UFF default values. The details of interaction parameters for the Lennard-Jones (12-6) potential and atomic partial charges obtained from the charge equilibration method are given in Table 1.
Table 1 Atomic partial charges and Lennard-Jones (12-6) parameters between atoms of the same type
2.3 MD simulations
MD simulations were performed using the Cerius Dynamics Simulation module. The simulation cells consisted of 2 ![]() 2
2 ![]() 2 unit cells for i-NiHCF. The simulations of water molecules inside the NiHCF thin films were performed at 300 K. The solid matrix atoms were fixed at the equilibrium lattice positions while the interstitial cations and water molecules were allowed to move during simulation. The simulations of bulk water were done on 832, 1 664 and 2 496 molecules respec- tively at a temperature of 300 K and a density of 0.998 g/cm3 in order to evaluate the suitability of the force fields. Then, aqueous solution simulations comprised of 832 water molecules and different combination of NaCl or CsCl (totally 15) were performed, corresponding to a molar concentration of 1.0 mol/L and a density from 1.056 7 g/cm3 for NaCl to 1.166 6 g/cm3 for CsCl at 300 K. The simulations of 1.0 mol/L solutions containing 5, 30 and 45 NaCl or CsCl were also performed. All simulations proceeded as follows. The system energy was first minimized by running the Cerius2 Minimizer module. Then, an MD simulation was performed using periodic boundary conditions(PBC) on the canonical N, V, T ensemble. The temperature was controlled using the Berendsen method of temperature-bath coupling (referred to as T-DAMPLING in Cerius2)[17]. The integration time step used in the simulation was 1
2 unit cells for i-NiHCF. The simulations of water molecules inside the NiHCF thin films were performed at 300 K. The solid matrix atoms were fixed at the equilibrium lattice positions while the interstitial cations and water molecules were allowed to move during simulation. The simulations of bulk water were done on 832, 1 664 and 2 496 molecules respec- tively at a temperature of 300 K and a density of 0.998 g/cm3 in order to evaluate the suitability of the force fields. Then, aqueous solution simulations comprised of 832 water molecules and different combination of NaCl or CsCl (totally 15) were performed, corresponding to a molar concentration of 1.0 mol/L and a density from 1.056 7 g/cm3 for NaCl to 1.166 6 g/cm3 for CsCl at 300 K. The simulations of 1.0 mol/L solutions containing 5, 30 and 45 NaCl or CsCl were also performed. All simulations proceeded as follows. The system energy was first minimized by running the Cerius2 Minimizer module. Then, an MD simulation was performed using periodic boundary conditions(PBC) on the canonical N, V, T ensemble. The temperature was controlled using the Berendsen method of temperature-bath coupling (referred to as T-DAMPLING in Cerius2)[17]. The integration time step used in the simulation was 1 ![]() 10-15 s. Unless otherwise stated, the configuration was equilibrated for 30 000 time steps, and the properties were obtained by averaging over another 30 000 time steps, using configurations saved every 100-time step.
10-15 s. Unless otherwise stated, the configuration was equilibrated for 30 000 time steps, and the properties were obtained by averaging over another 30 000 time steps, using configurations saved every 100-time step.
3 Results and discussion
3.1 Potential energy of reduced i-NiHCF unit cell
First we will investigate the various reduced i-NiHCF with different combinations of Na+ and Cs+. This is a defect structure, one iron and its coordinated cyano groups have been removed from each unit cell. Totally four cations (Na+ and Cs+) are added at the interstitial sites to balance the negative charge in these reduced matrices. Then QEq is used to recalculate the charge distribution on the matrix atoms and charge- compensating cations. Next, six water molecules are added per unit cell to satisfy the coordination sphere for the nickel atoms that lost CN- bonding[13]. The coordination sphere waters are present in every case discussed as follows, with additional hydration waters entering the structure to minimize the overall matrix energy. The defect structure i-NiHCF yields a nickel-rich unit cell, NaNi4[Fe(CN)6]3��6H2O) or CsNi4[Fe- (CN)6]3��6H2O) for the oxidized form and NaxCs4-xNi4- [Fe-(CN)6]3��6H2O) [0��x��4] for the reduced form, when Na and Cs are the intercalated cations. All water molecules have the same charges as in SPC/E model and thus do not cause a redistribution of charge within the solid matrix. Moreover, all of the intercalated water molecules are allowed to move during MD simulations. Varying the number of intercalated waters and minimizing the total potential energy of the matrices determine the energetically favored hydration states of the reduced matrices.
Fig.2 shows the dependence of potential energy (per unit cell) on the number of waters intercalated into the reduced i-NiHCF matrices with different combinations of Na+ and Cs+. For a fixed ratio of Na+/Cs+ in the reduced i-NiHCF, the potential energy is first decreased as water molecules are added to the basic unit cell. After the energy goes down to the lowest point, it is increased with the increasing of the number of water molecules. For various reduced i-NiHCF, the 4Na+/0Cs+ and 3Na+/ 1Cs+ combinations have energy minima with 16 added water molecules per unit cell (22 water molecules total, when counting the 6 coordination sphere waters); the 2Na+/2Cs+ and 1Na+/3Cs+ combinations have energy minima with 14 and the 0Na+/4Cs+ has energy minimum with 12 added water molecules per unit cell respectively. Also we can find in Fig.2 that the low energy parts of these curves become broader when the number of Cs+ in the matrices is increased. The reduced i-NiHCF with 4 intercalated Cs+ per unit cell has a broad energy minimum ranging from 8 to 14 added waters. For the same number of water molecules added in the matrices, the potential energy is increased with the increasing of the ratios of Cs+/Na+ in unit cell.
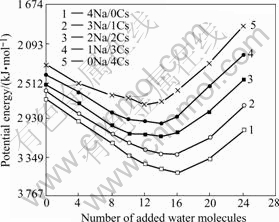
Fig.2 Unit cell potential energy with added waters as water molecules added to various reduced i-NiHCF
The potential energy at the minima energy states with the Cs+ proportion in unit cell is shown in Fig.3. The minimum energy grows linearly with Cs+ proportion y(Cs) when y(Cs) is lower than 0.5 or higher than 0.75; namely, the average potential energy will increase about 209.3 kJ/mol when one Na+ ion is replaced by one Cs+ ion in each unit cell. But it only increases about 125.6-
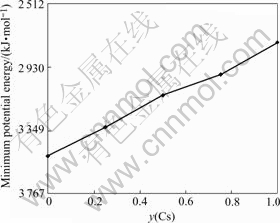
Fig.3 Potential energy at minimum energy states with Cs+ ratio in unit cell
142.3 kJ/mol when the ratio of Cs+ changes from 0.5 to 0.75.
3.2 Radial distribution function and configuration of i-NiHCF at equilibrium
The arrangement of water and cations inside various reduced i-NiHCF was further analyzed using radial distribution functions and configurationally snapshots in equilibrium. The sodium-oxygen radial distribution functions for reduced i-NiHCF with different combinations of Cs+/Na+ under the minimum energy condition are shown in Fig.4. For comparison, the sodium-oxygen radial distribution function for the bulk NaCl solution is also shown in Fig.4. The first gONa peaks for the 2Cs+/2Na+ and 3Cs+/1Na+ combinations in reduced i-NiHCF matrices have the same position as that for the bulk solution, while the ones for the 0Cs+/4Na+ and 1Cs+/3Na+ combinations move a little inward. This means that the average distance between sodium and oxygen inside the matrices in the reduced states for 2Cs+/2Na+ and 3Cs+/1Na+ combinations are the same as that in the bulk solution; while the average distance inside the matrices for 0Cs+/4Na+ and 1Cs+/3Na+ combinations are shorter than that in the bulk solution. These are understandable because there are 16 water molecules for 0Cs+/4Na+ and 1Cs+/3Na+ combinations but only 14 water molecules for 2Cs+/2Na+ and 3Cs+/1Na+ combinations in the same solid structure room at the minimum energy condition.
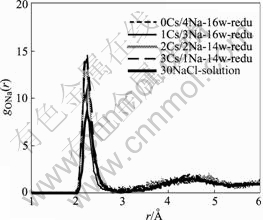
Fig.4 Radial distribution function of sodium-oxygen in various reduced i-NiHCF with different cation combinations
The equilibrium configuration snapshots obtained at the end of the simulation (30 ps) are shown in Fig.5 for two reduced matrices with 2Na+/2Cs+ and 1Na+/3Cs+ combinations at the minimum energy condition. In these configurations, the average distance between Na+ and oxygen is the same as that in the bulk solution, so these configurations are sterically favorable and thus energetically favorable too. But in 2Cs+/2Na+ combinations, four cations distribute at the four corners of the configuration; while in 3Cs+/1Na+, four cations mix well with intercalated waters. The configurational snapshot of 3Cs+/1Na+ combination shows substantial fluctuations in the number of waters and cations present in each unit cell, consistent with a liquid-like behavior.
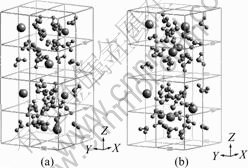
Fig.5 Snapshots of equilibrium configuration in reduced i-NiHCF with 2Cs+ /2Na+ combination(a) and with 3Cs+/1Na+ combination (b) loaded with appropriate amount of cation and water molecules (14 added water molecules for reduced per unit cell. Only two unit cells are shown for clarity.)
The cesium-oxygen radial distribution functions for the reduced states with 2Na+/2Cs+ and 1Na+/3Cs+ combinations at the minimum energy condition and for the bulk CsCl solution are shown in Fig.6. The first peaks appear at the same position for the reduced state with 1Na+/3Cs+ (14 added waters) and the bulk solution, but the first peak for 2Na+/2Cs+ combination moves inward. Thus, both the sodium-oxygen and the cesium-oxygen radial distribution functions show that, for the reduced states of i-NiHCF, 1Na+/3Cs+ combination is sterically favored.
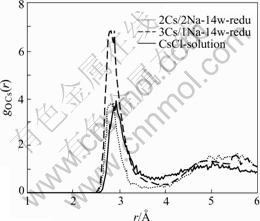
Fig.6 Radial distribution function of cesium-oxygen for reduced i-NiHCF
3.3 Aqueous potential energy of mixed Na/CsCl solution
The MD simulations of total 1 molar aqueous solution with different combination of NaCl and CsCl show that the potential energy of the solution will increase with the increasing of the Cs+ cation proportion in the bulk solution and it has a nearly linear relationship (not shown). The average potential energy change is about 167.4 kJ/mol when a Na+ cation is replaced with a Cs+ cation in 1 mol/L solutions. When ion exchange takes place between solution and solid in ESIX process, the solution will accept more Na+ cations in order to keep its minimum energy states. But we still can not determine the partitioning of Cs+ and Na+ into NiHCF thin film only according to the MD simulation results of solution or solid.
3.4 Total potential energy of solid and solution
The different distribution of cations in solution or solid will cause the change of total potential energy when the number of cations in solution and solid matrix is fixed. Because the minimum energy difference appears in potential energy curve when the Cs+ ratio y(Cs) changes from 0.5 to 0.75 in reduced i-NiHCF unit cell; solid structure transform from 2Cs+/2Na+ to 3Cs+/1Na+ combination needs one Cs+ ion in solution to replace one Na+ in solid, the solid potential energy will increase by 125.6-142.3 kJ/mol but the solution potential energy will decreases by 167.4 kJ/mol, so the total energy of the solution and the solid will decrease by about 25.12-41.86 kJ/mol. Similarly, when solid structure transforms from 3Cs+/1Na+ to 4Cs+/0Na+ combination, the total energy of the solution and the solid will increase by 25.12-41.86 kJ/mol. So combining the solid and solution simulation results we can find that the reduced i-NiHCF with the 3Cs+/1Na+ combination is the stablest state.
In all respects, the hydration results obtained for alkali-loaded i-NiHCF are sensible in light of the nature of the ion hydration in solution and the size of the interstitial site. The first peaks in gHNa and gHCs for bulk solutions show that the radii of the first hydration shells are approximately 3.7 and 4.5 ? (tail of the first peak) for Na+ and Cs+, respectively[10]. The average potential energies per water molecule for Na+ and Cs+ obtained from MD simulations are -126 and -55.7 kJ/mol, respectively[18], indicating the strong hydration of water to Na+. With a smaller hydration shell and larger hydration energy, Na+ cations prefer to be hydrated in the solution. The more Na+ exists in 1 mol/L mixed solution, the lower the potential energy of the whole solution will be. Cs+ has a bigger hydration shell and lower hydration energy, so it is easier to get into the solid in ESIX experiment than Na+. The i-NiHCF can provide a big interstitial site for one Cs+ so the oxidized i-NiHCF matrix displays a high Cs+ selectivity for all solution compositions examined in ESIX experiment. Though the size of the Cs+ is bigger than that of Na+, the interstitial sites within reduced i-NiHCF are large enough to accommodate 3Cs+/1Na+ and water without dramatically altering the hydration distance. However, when four big Cs+ cations are intercalated into the reduced state, the interstitial sites are not large enough to readily permit a hydrated Cs+ and strong repulsive Cs+-Cs+ interactions will happen.
4 Conclusions
Molecular dynamics simulations have been done when different amounts of water molecules and different proportions of Cs+ and Na+ are intercalated into the reduced i-NiHCF matrix. For the fixed proportion of cations, the potential energy of the structure is first decreased with increasing water number, and then is increased after reaching the lowest point of the energy. Both the solid energy and the solution energy are increased when the Cs+ proportion is increased. The minimum energy differences of different composition Cs+ and Na+ in i-NiHCF occurs when the ratio of Cs+/Na+ transits from 2:2 to 3:1. The simulation results combining the solid and solution show that strong repulsive Cs+-Cs+ interactions exist in the solid and give rise to a stable solid composition possessing 75% Cs+ and 25% Na+. These are consistent with the experimental observation[7, 10] over a range of solution compositions.
Acknowledgment
The authors gratefully acknowledge support provided by U.S. National Science Foundation grant No. CTS-0236608.
References
[1] BOCARSLY A B, SINHA S. Effects of surface structure on electrode charge transfer properties: Induction of ion selectivity at the chemically derivatized interface [J]. J Electroanal Chem, 1982, 140(1): 167-172.
[2] BACSKAI J, MARTINUSZ K, CZIROK E, INZELT G, KULESZA P J, MALIK M A. Polynuclear nickel hexacyanoferrates: monitoring of film growth and hydrated counter-cation flux/storage during redox reactions [J]. J Electroanal Chem, 1995, 385: 241-248.
[3] STEEN W A, SCHWARTZ D T. A route to diverse combinatorial libraries of electroactive nickel hexacyanoferrate [J]. Chem Mater, 2003, 15(12): 2449-2453.
[4] HAO X, SCHWARTZ D T. Tuning intercalation sites in nickel hexacyanoferrate using lattice nonstoichiometry [J]. Chem Mater, 2005, 17(23): 5831-5836.
[5] HAO X, GUO J, LIU S, SUN Y. Electrochemically switched ion exchange performances of capillary deposited nickel hexacyano- ferrate thin films [J]. Trans Nonferrous Met Soc China, 2006, 16(3): 556-561..
[6] RASSAT S D, SUKAMTO J H, ORTH R J, LILGA M A, HALLEN R T. Development of an electrically switched ion exchange process for selective ion separations [J]. Sep Purif Technol, 1999, 15: 207-222.
[7] JEERAGE K M, SCHWARTZ D T. Characterization of cathodically deposited nickel hexacyanoferrate for electrochemically switched ion exchange [J]. Sep Sci Technol, 2000, 35(15): 2375-2392.
[8] STEEN W A, HAN S, YU Q, GORDON R A, CROSS J O, STERN E A, SEIDLER G T, JEERAGE K M, SCHWARTZ D T. Structure of cathodically deposited nickel hexacyanoferrate thin films using XRD and EXAFS [J]. Langmuir, 2002, 18: 7714-7721.
[9] STEEN W A, JEERAGE K M, SCHWARTZ D T. Raman spectroscopy of redox activity in cathodically electrodeposited nickel hexacyanoferrate thin films [J]. Appl Spectrosc, 2002, 56(8): 1021-1029.
[10] JEERAGE K M, STEEN W A, SCHWARTZ D T. Charge- density-dependent partitioning of Cs+ and K+ into nickel hexacyanoferrate matrixes [J]. Langmuir, 2002, 18: 3620-3625.
[11] JEERAGE K M, STEEN W A, SCHWARTZ D T. Correlating nanoscale structure with ion intercalation in electrodeposited nickel hexacyanoferrate thin films [J]. Chem Mater, 2002, 14: 530-535.
[12] YU Q, STEEN W A, JEERAGE K M, JIANG S, SCHWARTZ D T. Structure-dependent solvent and ion intercalation in reduced and oxidized nickel hexacyanoferrates [J]. J Electrochem Soc, 2002, 149(6): E195-E203.
[13] KAWAMOTO T, ASAI Y, ABE S. Ab initio calculations on the mechanism of charge transfer in Co-Fe Prussian-blue compounds [J]. Phys Rev B, 1999, 60: 12990-12993.
[14] RAPPE A K, GODDARD W A III. Charge equilibration for molecular dynamics simulations [J]. J Phys Chem, 1991, 95: 3358-3363.
[15] RAPPE A K, CASEWIT C J, COLWELL K S, GODDARD W A, SKIFF W M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations [J]. J Am Chem Soc, 1992, 114: 10024-10035.
[16] BERENDSEN H J C, GRIGERA J R, STRAATSMA T P. The missing term in effective pair potentials [J]. J Phys Chem, 1987, 91: 6269-6271.
[17] BERENDSEN H J C, POSTMA J P M, VAN GUNSTEREN W F, DINOLA A, HAAK J R. Molecular dynamics with coupling to an external bath [J]. J Chem Phys, 1984, 81: 3684-3690.
[18] LEE S H, RASAIAH J C. Molecular dynamics simulation of ion mobility. 2. alkali metal and halide ions using the SPC/E model for water at 25 �� [J]. J Phys Chem, 1996, 100: 1420-1425.
(Edited by YUAN Sai-qian)
Foundation item: Project (20006011) supported by the National Natural Science Foundation of China; Project (20021017) by the Natural Science Foundation of Shanxi Province; Project (2004-24) by the Scholarship Council Foundation of Shanxi Province
Corresponding author: HAO Xiao-gang; Tel: +86-351-6018193; Fax: +86-351-6018554; E-mail: xghao@tyut.edu.cn
