
J. Cent. South Univ. Technol. (2010) 17: 943-946
DOI: 10.1007/s11771-010-0581-6
![]()
Light energy conversion device for photocatalyst 2.0%WO3-TiO2 withoxygen vacancies for water splitting
TONG Hai-xia(ͯ��ϼ)1, CHEN Qi-yuan(����Ԫ)2, HU Hui-ping(����Ƽ)2, YIN Zhou-lan(������)2
1. Institute of Chemical and Biologic Engineering, Changsha University of Science and Technology,
Changsha 410076, China;
2. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
? Central South University Press and Springer-Verlag Berlin Heidelberg 2010
Abstract:
Using carbon felt, polytetrafluoroethylene latex and powder catalyst to assembly a light energy conversion device, the photocatalytic activity of catalyst 2.0%WO3-TiO2 (2%WO3 compounding TiO2) with oxygen vacancies was studied through the water splitting for O2 evolution, using a high pressure mercury lamp as the light source and Fe3+ as the electron acceptor in two different devices: an ordinary photolysis device with catalyst powder suspending through a magnetic stirrer and a self-assembly light energy conversion device. The results show that after 12 h irradiation, the photocatalytic activity of 2.0%WO3-TiO2 with oxygen vacancies in the self-assembly light energy conversion device is higher than that of the ordinary photolysis device, and the amount of oxygen evolution is about 12 and 9 mmol/L respectively in these two devices. After 12 h, the rates of O2 evolution are slow in each device and the photocatalyst almost loses the photoactivity in the ordinary photolysis device. So, compared with the ordinary photocatalytic device, the rate of oxygen evolution and the life time of the catalyst are improved in the self-assembly light energy conversion device.
Key words:
1 Introduction
With the gradual depletion of fossil energy, more and more attention is paid to renewable hydrogen energy [1-7]. The key to the development of hydrogen energy is to produce hydrogen at a large-scale and low-cost. The main existing technologies for hydrogen production are as follows [8]: (1) electrolysis water to produce hydrogen, which will consume electric energy, and most of which is from fossil fuels; (2) using biomass to produce hydrogen energy, which is a way to obtain low-cost hydrogen energy; (3) photo splitting water to produce hydrogen energy, which is the main way to obtain hydrogen on a large-scale and at low-cost, which is regarded as the most prospective hydrogen producing technology in the 21st century. There are three paths to achieve photo splitting water [9]: (1) electrochemical decomposition of water to produce hydrogen through photo-electro-chemical pool. The solar energy is converted into electric energy through the photoelectrode and then electrolyzed water into H2 and O2 from the anode and cathode, respectively. It is easy to realize the decomposition of water in this way but there is a low energy conservation efficiency because the solar energy should firstly be converted into electric energy to electrolyze water to produce H2; (2) photo-complexing decomposition of water to produce H2. Using transition metals such as porphyrin as a photosensitizer to decompose water into H2 and O2 through false photosynthesis, and this process simulates the photosynthesis of plants. However, photosensitizers are so expensive and the catalysts are easy to loss their activities, which results in high cost of H2; and (3) photo splitting water by semiconductor photocatalysts. A series of semiconductor photocatalysts such as TiO2 catalytically split water molecules into hydrogen molecules and oxygen molecules through the conversion of solar energy. This method is environmental friendly, and of great strategic significance for the development of new energy sources. In the experimental procedure, magnetic stirrer must be used to keep the catalyst powder suspended, which causes the increase of the cost of H2 evolution. In 2007, MASAAKI et al [10] produced H2 and O2 by photocatalytic splitting water using Pt-loaded visible light-responsive TiO2 thin film photocatalysts.
The thin film photocatalytic device consisted of two parts: one was a Ti foil substrate deposited with a TiO2 thin film on one side, which was immersed in 1.0 mol/L NaOH and Pt particle (0.68 mg/cm2) on the other side, which was immersed in 0.5 mol/L H2SO4 aqueous solution in order to add a small chemical bias to assist the electron side to the Pt side through the metal substrate; the other is a Nafion film which was mounted at the center of an H-type glass container. After irradiation for 15 h, the evolution of H2 and O2 observed was about 1.2 and 0.6 ?mol, respectively. In order to improve the rate of gas evolution, MASAYA et al [11] modified the catalysts and the rates of H2 and O2 evolution were increased greatly to 80 and 40 ?mol within 7 h irradiation, respectively.
In our research group the reaction of photocatalytic splitting water into H2 and O2 was carried out via two half reactions in the ��Double-Bed�� splitting water system. One is the photo-reduction with H2 evolution and the other is the photo-oxidation with O2 evolution using Cr-K2La2Ti3O10 [12] and 2.0% WO3-TiO2 [13] as catalysts, respectively. At last, the two half reactions were connected in the light energy conversion device. In this work, the half reaction of the photo-oxidation with O2 evolution was investigated using Fe3+ as electron acceptors and 2% WO3-TiO2 with oxygen vacancies as catalysts.
2 Theory of water photolysis device
From the chemical thermodynamics, under the standard state (101.325 kPa, 298.15 K) the change in normal mole Gibbs function![]() of the reaction H2(g)+1/2O2(g)=H2O(l) is -237.19 kJ/mol��0, which means that the reaction can occur automatically. The reaction of the water formation can be designed to cell��:
of the reaction H2(g)+1/2O2(g)=H2O(l) is -237.19 kJ/mol��0, which means that the reaction can occur automatically. The reaction of the water formation can be designed to cell��:
-)Pt | H2(p��) | H+(a=1) | O2(p��) | Pt(+Anode reaction: 1/2O2+2H++2e=H2O
Cathod reaction: H2-2e=2H+Cell reaction: H2 (p��)+1/2O2 (p��)=H2O (l)
At 298.15 K, the electrical potential difference between the anode and the cathode is 1.23 V. The ![]() (298.15 K, p��) of the reaction of water decomposition into H2 and O2: H2O(l)=H2(g)+1/2O2(g) is 237.19 kJ/mol ��0, indicating that the reaction cannot occur automatically. ?rGT��=246 856-55.96 T (J/mol) of reaction H2O(g)=H2(g)+1/2O2(g) is 246.8 kJ/mol [14], so it needs a very high temperature to split water automatically. The reaction of the water decomposition can be designed to cell ��:
(298.15 K, p��) of the reaction of water decomposition into H2 and O2: H2O(l)=H2(g)+1/2O2(g) is 237.19 kJ/mol ��0, indicating that the reaction cannot occur automatically. ?rGT��=246 856-55.96 T (J/mol) of reaction H2O(g)=H2(g)+1/2O2(g) is 246.8 kJ/mol [14], so it needs a very high temperature to split water automatically. The reaction of the water decomposition can be designed to cell ��:
��)Pt | O2(p��) | H+(a=1) | H2(p��) | Pt(+Anode reaction: 2H++2e=H2
Cathode reaction: H2O-2e=1/2O2+2H+Cell reaction: H2O (l) =H2 (p��)+1/2O2 (p��)
At 298.15 K, the reaction of H2 evolution takes place on the anode of the cell, and the reaction of O2 evolution takes place on the cathode of the cell with the electrical potential difference being 1.23 V. That is, the electromotive force of the cell is ![]() -1.23 V.
-1.23 V.
If the electromotive force of the cell can be changed into positive value (![]() ��0 ) the reaction of the cell �� will occur, automatically, that is, the decomposition of water H2O(l) =H2(g)+1/2O2(g) will take place automatically. Due to the effect of light, the required balance potential of the illuminated semiconductor electrodes is lower than that of the unilluminated semiconductor electrode in the electrochemical reaction. So, if a suitable semiconductor is chosen as the electrode of cell ��, the electric potential for H2 evolution will change more positively, and the electric potential for O2 evolution will change more negatively after illuminated. When the electric potential for H2 evolution is more positive than that for O2 evolution, and the electrical were potential difference of cell �� changes into a positive value (
��0 ) the reaction of the cell �� will occur, automatically, that is, the decomposition of water H2O(l) =H2(g)+1/2O2(g) will take place automatically. Due to the effect of light, the required balance potential of the illuminated semiconductor electrodes is lower than that of the unilluminated semiconductor electrode in the electrochemical reaction. So, if a suitable semiconductor is chosen as the electrode of cell ��, the electric potential for H2 evolution will change more positively, and the electric potential for O2 evolution will change more negatively after illuminated. When the electric potential for H2 evolution is more positive than that for O2 evolution, and the electrical were potential difference of cell �� changes into a positive value (![]() ��0), the decomposition of water H2O(l)= H2(g)+1/2O2(g) will take place automatically. The schematic diagram of cell �� for photo splitting water with H2 and O2 evolution is shown in Fig.1.
��0), the decomposition of water H2O(l)= H2(g)+1/2O2(g) will take place automatically. The schematic diagram of cell �� for photo splitting water with H2 and O2 evolution is shown in Fig.1.
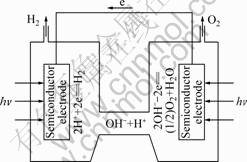
Fig.1 Schematic diagram of cell �� for photo splitting water with H2 and O2 evolution
The shortcomings and the solutions of the semiconductor particles suspended in the water for the water photolysis reaction are as follows.
(1) To keep powder particles suspended in water photolysis device a stirrer should be used, which has to consume energy.
(2) It is difficult to separate powder particles from the solution. So, when renewing the solution of electron acceptors or electron donors, the reaction should be stopped. Therefore, the catalyst powder is fixed and prepared as the semiconductor electrode to address the above-mentioned powder suspension with the issue of water photolysis and at the same time to ensure the catalytic activity of the materials. It is convenient to make use of light energy to fix catalyst powder as semiconductor electrode. The device of water splitting can be built as an electrochemical reactor just like cell ��, and even the electron acceptors or electron donors can be replaced by the electron transfer between the photoelectrodes in the device.
3 Experimental
3.1 Fabrication of 2.0%WO3-TiO2 photoelectrode
Because of its large porosity, good electrical conductivity and irregular three-dimensional network structure, the carbon felt composed of carbon fibers was chosen as the vector for the fixing of the catalyst powder 2.0%WO3-TiO2 with oxygen vacancies. The detailed steps are as follows.
(1) Each side of the carbon felt (3 cm��14 cm�� 18 cm) was coated with a thin blanket of polytetrafluoroethylene latex and then put on the electric oven and thermally treated for 0.5 h at about 100 ��. Thus, the polytetrafluoroetheylene latex was solidified, and the carbon felt was coated and filmed.
(2) The catalyst powder 2.0%WO3-TiO2 with oxygen vacancies was dispersed evenly on the side of the carbon felt processed as step (1). Then, the carbon felt was loaded with the catalyst powder and rolled into a cylinder which was then stitched. So, TiO2 photocathode was finished.
3.2 Light energy conversion device
The ordinary photolysis device is shown in Fig.2 [16]. In the device magnetic stirrer was used to keep the
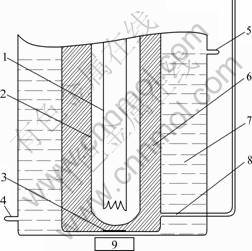
Fig.2 Sketch map of ordinary photocatalytic reactor: 1��Hg lamp; 2��Glass jacket; 3��Magnetic bar; 4��Cooling water inlet; 5��Cooling water outlet; 6��Mixture of reactor; 7�� Cooling water; 8��Outlet; 9��Magnetic stirrer
catalyst powder suspended. However, in the self- assembly light energy conversion device (shown in Fig.3) the catalyst powder was fixed and prepared as semiconductor photo electrode, and no additional electric energy was consumed to keep it suspended in the catalytic procedure. The carbon felt in the self-assembly device acted as an electrode. The reaction formed a primary cell during the process of photo reaction in our self-assembly light energy conversion device as follows:-)C|2.0%WO3-TiO2 with oxygen vacancies, 2.0%WO3-TiO2 with oxygen vacancies (h+, e-), H2O, O2, H+, Fe3+, Fe2+�OC (+,Because of the conductivity of the carbon felt, the distance of the photo-electrons and the holes is enlarged, which hinders their recombination. The photo catalytic activity of 2.0%WO3-TiO2 with oxygen vacancies was improved.
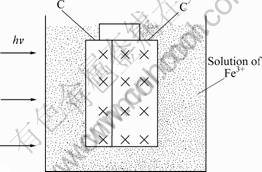
Fig.3 Sketch map of light energy conversion device
4 Results and discussion
Compared with the ordinary photolysis device, the self-assembly light energy conversion device becomes simpler and higher-energy efficient because the magnetic stirrer is no longer required to keep the catalyst powder suspended in the self-assembly light energy conversion device.
The photocatalytic activities of 2.0%WO3-TiO2 with oxygen vacancies were studied through the splitting water for O2 evolution under UV-light irradiation using Fe3+ as electron acceptors in the ordinary device and the self-assembly device, respectively. The results are presented in Figs.4 and 5. After 12 h irradiation the amount of oxygen is about 12 and 9 mmol/L respectively in these two devices, and the average rate of O2 evolution in the ordinary photolysis device is larger than that in the ordinary photolysis device. From Fig.5 in the self-assembly device the largest amount for O2 evolution is about 2.295 mmol/L at the 4th hour irradiation and 1.699 mmol/L in the ordinary device at the 9th hour. After 12 h irradiation, the rates of O2 evolution are slow in each device, and the photo catalyst almost loses the photoactivity in the ordinary photolysis device. So the life time of the catalyst in the light energy conversion device is a little longer than that in the ordinary photocatalytic device.
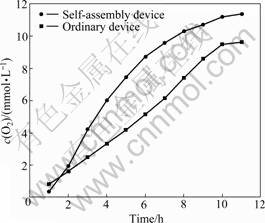
Fig.4 Catalytic activities of catalyst 2%WO3-TiO2 with oxygen vacancies in ordinary device and self-assembly device
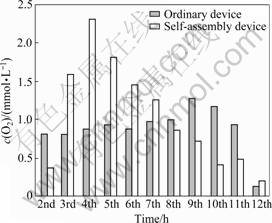
Fig.5 Comparison of rates of O2 evolution in ordinary device and self-assembly device in each hour
5 Conclusions
(1) The catalyst 2.0%WO3-TiO2 with oxygen vacancies is attached in the carbon felt, and used to make semiconductor photoelectrode under the effect of polytetrafluoroethylene. The problem of keeping the catalyst powder suspended through the use of outside energy is solved.
(2) After 12 h irradiation the amount of oxygen is about 12 and 9 mmol/L, respectively in the self-assembly light energy conversion device. The rates of O2 evolution are slow in each device and the photo catalyst almost loses the photoactivity in the ordinary photolysis device.
(3) Compared with that in the ordinary photocatalytic device, the rate of oxygen evolution and the life time of the catalyst are improved in the self-assembly light energy conversion device.
References
[1] ZONG X, YAN H J, WU G P, MA G J, WEN F Y, WANG L, LI C. Enhancement of photocatalytic H2 evolution on CdS by loading MoS2 as cocatalyst under visible light irradiation [J]. J Am Chem Soc, 2008, 130(23): 7176-7180.
[2] CHEN Hai-qun, ZHU Jun-wu, MA Jun-xing, PANG Kai, SUN Xiao-qiang. Synthesis of visible light responsive ultrafine K4Ce2Nb10O30 by a stearic acid method [J]. J Rare Earths, 2009, 27(5): 811-814.
[3] BAMWENDA G R, UESIGI T, ABE Y, SAYAMA K, ARAKAWA H. The photocatalytic oxidation of water to O2 over pure CeO2, WO3 and TiO2 using Fe3+ and Ce4+ as electron acceptors [J]. Applied Catalysis A: General, 2001, 205: 117-128.
[4] BAMWENDA G R, ARAKAWA H. Cerium dioxide as a photocatalyst for water decomposition to O2 in the presence of Ceaq+4 and Feaq+3 species [J]. Journal of Molecular Catalysis A: Chemical, 2000, 161(1/2): 105-113.
[5] ABE R, SAYAMA K, ARAKAWA H. Significant effect of iodide addition on water splitting into H2 and O2 over Pt loaded TiO2 photocatalyst: Suppression of backward reaction [J]. Chemical Physics Letters, 2003, 371(3/4): 360-364.
[6] ZOU Z G, ARAKAWA H. Direct water splitting into H2 and O2 under visible light irradiation with a new series of mixed semiconductor photocatalyst [J]. Journal of Photochemistry and Photobiology A: Chemistry, 2003, 158(2/3): 145-162.
[7] ZOU Z G, YE J H, ARAKAWA H. Photophysical and photocatalytic properties of InMO4(M=Nb5+,Ta5+) under visible light irradiation [J]. Materials Research Bulletin, 2001, 36: 1185-1193.
[8] CHEN Qi-yuan, YANG Ya-hui, YIN Zhou-lan, LI Jie. Practical hydrogen production from water splitting analysis of the technical prospects [M]. Beijing: Science Press, 2005: 379-387. (in Chinese)
[9] LI Xin-yong, L? Gong-xuan, LI Shu-ben. Progress of semiconductor research in photolysis of water [J]. Molecular Catalysis, 2001, 15(1): 72-79.
[10] MASAAKI K, MASATO T, MASAYA M, JOHN M T, MASAKAZU A. Photocatalytic water splitting using Pt-loaded visible light-responsive TiO2 thin film photocatalysts [J]. Catal Today, 2007, 120: 133-138.
[11] MASAYA M, MASAAKI K, SHOHEI F, KAZUSHI I, MASATO T, MASAKAZU A. The effect of the hydrothermal treatment with aqueous NaOH solution on the photocatalytic and photoelectrochemical properties of visible light-responsive TiO2 thin films [J]. Catal Today, 2008, 132: 159-164.
[12] UCHIDA S, YAMAMOTO Y, FUJISHIRO Y. Intercalation of titanium oxide in layered H2Ti4O9 and H4Nb6O17 and photocatalytic water cleavage with H2Ti4O9/(TiO2, Pt) and H4Nb6O17/(TiO2, Pt) nanocomposites [J]. J Chem Soc: Faraday Tans, 1997, 93(17): 3229- 3234.
[13] TONG Hai-xia. The preparation of modified rutile TiO2 catalysts and the study of their photocatalytic activity for water splitting with O2 evolution [D]. Changsha: Central South University, 2008. (in Chinese)
[14] HUANG Xing-wu. Principle of non-ferrous metallurgy [M]. Beijing: Metallurgical Industry Press, 1993: 30.
[15] TONG Hai-xia, CHEN Qi-yuan, YIN Zhou-lan, HU Hui-ping, WU Dao-xin, YANG Ya-hui. Preparation, characterization and photo-catalytic behavior of WO3-TiO2 catalysts with oxygen vacancies [J]. Transactions of Nonferrous Metals Society of China, 2009, 19(6): 1483-1488.
[16] TONG Hai-xia, CHEN Qi-yuan, YIN Zhou-lan, LI Jie, HU Hui-ping. Preparation of TiO2/Nb2O5 photocatalyst loaded with WO3 for photocatalytic oxidation of water with O2 evolution [J]. Journal of Central South University of Technology, 2007, 14(6): 788-792.
Foundation item: Project(2010CL04) supported by the Hunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, China; Project(K-081025) supported by State Key Laboratory Breeding Base of Photocatalysis, Fuzhou University, China
Received date: 2010-01-12; Accepted date: 2010-04-08
Corresponding author: TONG Hai-xia, PhD; Tel: +86-731-85258733; E-mail: tonghaixia@126.com
